причины, симптомы, диагностика, лечение в НКЦ ОАО «РЖД», с филиалом ЦКБ № 1
Амилоидоз почек — заболевание нарушения обмена веществ, в результате чего в тканях образуется субстанция — амилоид, которая нарушает функций органов.
Причины амилоидоза почек:
Данная болезнь, как правило, возникает после длительного хронического воспаления (туберкулеза, сифилиса), при ревматоидном артрите, хронических нагноениях (остеомиелит), опухолях и других заболеваниях.
Амилоидные субстанции часто откладываются в почках, нервной системе, сердце и кишечнике. Первичную форму болезни врачи связывают именно с наследственными причинами.
Если вам поставлен диагноз амилоидоз почек, лечение следует начинать незамедлительно. Это позволит избежать осложнений, в том числе и на сердце, так как накопление амилоида в сердечных тканях приводит к развитию сердечной недостаточности.
Симптомы и диагностика амилоидоза:
Амилоидоз, согласно медицинской статистике, встречается часто. Больше всего при данном заболевании поражаются почки. Локализованные формы амилоидоза кожи или мочевого пузыря могут протекать бессимптомно долгое время. Заболевание затем проявляется в виде отеков, общей слабости, развития почечной недостаточности, перепадами артериального давления, иногда диареей. В ряде случаев больные жалуются на одышку и аритмию, нарушения чувствительности нервных окончаний.
При диагностике в моче пациента обнаруживается белок. Длительная потеря белка в сочетании с другими факторов приводят к развитию гипопротеинемии (снижению белка в крови) и связанного с ней отека.
Биопсия почки, слизистой оболочки прямой кишки, либо ткани десны сегодня является самым достоверным методом диагностики.
В нефрологическом отделении нашей клиники можно пройти всестороннее обследование, в том числе биопсию почки.
Амилоидоз почек. Лечение
После постановки пациенту точного диагноза «амилоидоз почек», проводится системное лечение. Мы накопили богатый опыт по профилактике и лечению пациентов с такими диагнозами, как амилоидоз кожи, мочевого пузыря, и т.д.
Врачи индивидуально подберут тип лечения: диетические программы, препараты с учётом сопутствующей патологии. Лечение амилоидоза почек проводится совместно со специалистами-кардиологами, гастроэнтерологами и неврологами.

Лечение амилоидоза почек — врачи, лечащие заболевание
Нефрологи Москвы — последние отзывы
Очень замечательный врач и лучший невролог, которого я когда — либо встречала. Она провела полное обследование, направила к другим специалистам и назначила мне лечение. Очень квалифицированный и внимательный доктор.
Ирина, 07 апреля 2021
Очень вежливый, спокойный, компетентный, располагающий к себе и любезный доктор. Мне было очень комфортно ему рассказывать о своих проблемах со здоровьем. Врач все со мной обсудил, осмотрел меня, посмотрел предыдущие результаты обследования и дал направление на анализы и памятку о том, как их правильно сдавать. Я сдам анализы и через несколько дней к нему вернусь. Этого специалиста мне порекомендовали.
Мне было очень комфортно ему рассказывать о своих проблемах со здоровьем. Врач все со мной обсудил, осмотрел меня, посмотрел предыдущие результаты обследования и дал направление на анализы и памятку о том, как их правильно сдавать. Я сдам анализы и через несколько дней к нему вернусь. Этого специалиста мне порекомендовали.
Аида, 28 марта 2021
Профессиональный, позитивный, высококвалифицированный, внимательный, отзывчивый и приятный в общении доктор. Она собрала анамнез, задала мне вопросы, назначила исследование и проанализировала его.
Наталья, 15 марта 2021
На модерации, 16 апреля 2021
Хочу вернуть время обратно и не сходить к этому врачу, ехала с мытищ 3 часа в пробках на такси, как толька зашла она мне два раза сказала что кроме того что нефролог она ещё заместитель глав врача, как будто мне это интересно было.
Даяна, 14 апреля 2021
Хороший и квалифицированный врач. Он нас принял, все рассказал, сделал УЗИ и дал рекомендации.
Вячеслав, 09 апреля 2021
 Она меня проконсультировала по поводу почек и обстоятельно все объяснила.
Она меня проконсультировала по поводу почек и обстоятельно все объяснила.Надежда, 09 апреля 2021
Александр, 07 апреля 2021
Очень внимательный врач.
Ольга, 07 апреля 2021
Доктор как профессионал соответствует своему уровню и как человек очень приятный в общении. Она просмотрела результаты анализов, осмотрела пациента, всё объяснила и проконсультировала нас.
Татьяна, 06 апреля 2021
Показать 10 отзывов из 1262Амилоидоз почек — симптомы, причины появления, диагностика, лечение
Амилоидоз почечной ткани — это патологическое состояние, которое сопровождается нарушением белкового и других видов обменных процессов в организме человека. В процессе заболеваемости в тканях органов и тканей, включая почечную паренхиму, формируется амилоид.
В процессе заболеваемости в тканях органов и тканей, включая почечную паренхиму, формируется амилоид.
Амилоид — это комплексная субстанция, которая состоит из белков и полисахаридов, скопление в органах данного вещества способствует угнетению из деятельности.
Амилоидоз почек и других паренхиматозных органов, развивается на фоне хронического воспалительного процесса в организме, к которым относятся сифилис и туберкулёзная инфекция. Также возможно формирование патологии при ревматических системных поражениях, остеомиелите и онкологических образованиях.
Симптоматические признаки на начальной латентной стадии могут и не проявляться, предположить развитие данной патологии можно только на основании жалоб больного, анамнеза развития заболевания, а также по результатам лабораторных показателей крови и мочи. В качестве дополнительных методов исследования больным в обязательном порядке должно быть проведено биопсионное исследование почечной паренхимы, компьютерная томография и экскреторная урография.
Указанные методы дополнительной диагностики поспособствую определению локализации патологического процесса и определения степени нарушения функционирования почечной паренхимы.
Основные методики проведения лечебных мероприятий
Наиболее важным моментом в проведении лечебных мероприятий является проведение этиотропной терапии. Этиотропное лечение подразумевает лечение инфекционных заболеваний, которые спровоцировали развитие заболевания. Очень часто при полном излечении от основной патологии, устраняются симптомы амилоидоза.
Специфическая терапия амилоидоза должна проводиться следующими средствами:
1. Ежедневное соблюдение режима диетического питания, которое подразумевает в себе полное или частичное ограничение употребления поваренной соли. Повышение в суточном рационе углеводов и витаминов.
2. Проведение патогенетического лечения с помощью такого препарата как хлорохин, и применение лекарственных средств, которые обладают десенсибилизирующими свойствами.
3. Симптоматический этап проведения лечения подразумевает назначение диуретических, гипотензивных лекарственных средств. При амилоидозе возможно использование трансфузии плазматической донорской жидкости и альбуминов.
При запущенной форме заболевания и тяжёлом общем состоянии человека показано проведении процедуры гемодиализа, такие больные становятся в очереди по трансплантации почки.
Прогноз на выздоровление у больных с амилоидозом полностью зависит от того насколько своевременно и правильно проводилось лечение. Не маловажный момент в построении прогноза на выздоровление принадлежит скорость прогрессирования заболевания. При обнаружении каких-либо изменений со стороны мочевыделительной системы следует обращаться к урологу или нефрологу для своевременного обнаружения и лечения заболеваний.
Получить консультацию
врача-терапевта
современные методы диагностики и лечения uMEDp
В статье дан обзор современных представлений о классификации, патогенезе, клинических проявлениях, диагностике и подходах к лечению системного амилоидоза. Наиболее подробно обсуждены проблемы первичного AL-, вторичного АА- и диализного А-бета-2-М-амилоидоза.
Наиболее подробно обсуждены проблемы первичного AL-, вторичного АА- и диализного А-бета-2-М-амилоидоза.
Таблица. Классификация амилоидоза
Термин «амилоидоз» объединяет заболевания, которые характеризуются внеклеточным отложением специфического нерастворимого фибриллярного белка амилоида. Немецкий патолог Рудольф Вирхов в 1853 г. предложил термин «амилоид» для обозначения вещества, откладывающегося в органах больных «сальной болезнью» (как называли амилоидоз в начале XIX века) при туберкулезе, сифилисе, лепре. Это вещество он ошибочно посчитал крахмалоподобным из-за характерной реакции с иодом. В настоящее время установлено, что полисахариды составляют не более 4% от массы амилоида, однако термины «амилоид» и «амилоидоз» закрепились.
Эпидемиология амилоидоза
Распространенность амилоидоза до настоящего времени изучена недостаточно. По данным S.Y. Tan и соавт. (1995) [1], реактивный АА-амилоидоз, один из наиболее частых вариантов амилоидоза, развивается у 5% больных с хроническими воспалительными заболеваниями в Европе; по данным других источников, АА-амилоидоз осложняет течение ревматоидного артрита в 6–10% случаев. В среднем доля АА-амилоидной нефропатии в структуре заболеваний почек в Европе составляет 2,5–2,8%, а в структуре болезней, приведших к хронической почечной недостаточности (ХПН), – 1% (в соответствии с данными Европейской ассоциации диализа и трансплантации).
(1995) [1], реактивный АА-амилоидоз, один из наиболее частых вариантов амилоидоза, развивается у 5% больных с хроническими воспалительными заболеваниями в Европе; по данным других источников, АА-амилоидоз осложняет течение ревматоидного артрита в 6–10% случаев. В среднем доля АА-амилоидной нефропатии в структуре заболеваний почек в Европе составляет 2,5–2,8%, а в структуре болезней, приведших к хронической почечной недостаточности (ХПН), – 1% (в соответствии с данными Европейской ассоциации диализа и трансплантации).
В США частота амилоидоза варьирует от 5,1 до 12,8 случаев на 100 тыс. населения в год [2]. Эти данные касаются преимущественно распространенности первичного AL-амилоидоза, или амилоидоза в рамках миеломной болезни и других В-гемобластозов. В странах третьего мира, по мнению S.Y. Tan (1995), смертность от AL-амилоидоза составляет 1 на 2000 населения (0,05%). По-видимому, представленные данные о распространенности различных вариантов амилоидоза в целом могут быть экстраполированы и на другие страны.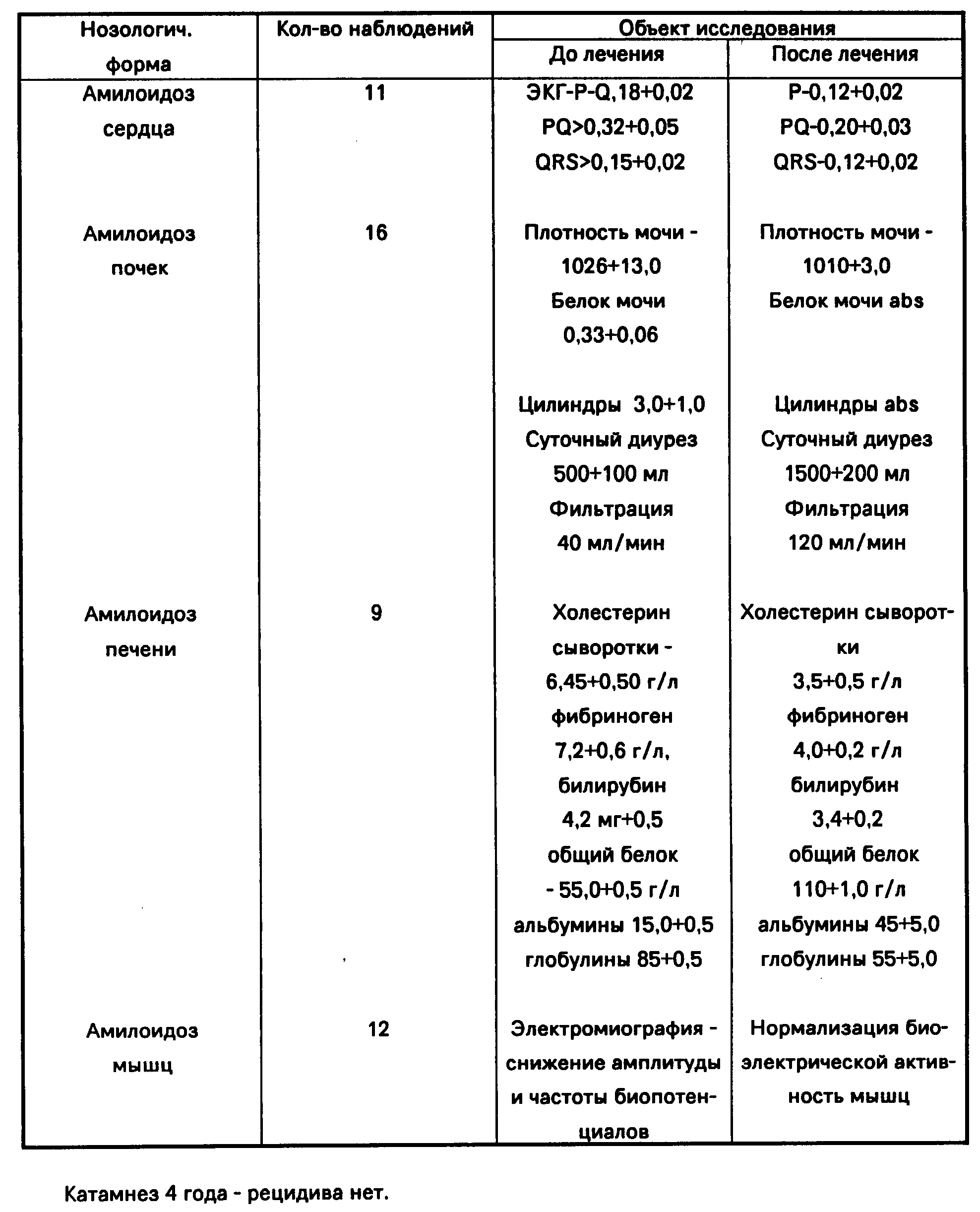
Морфология амилоидоза
Основу тканевых отложений амилоида составляют амилоидные фибриллы – особые белковые структуры диаметром 5–10 нм и длиной до 800 нм, состоящие из 2 и более параллельных разнонаправленных (антипараллельных) филаментов, образующих кросс-бета-складчатую конформацию. Именно она определяет специфическое оптическое свойство амилоида – способность к двойному лучепреломлению. При микроскопии окрашенных конго красным препаратов в поляризованном свете амилоид изменяет красный цвет окраски на яблочно-зеленое свечение. Обнаружение этого свойства положено в основу диагностики амилоидоза.
Структурные и химико-физические особенности амилоида определяются основным белком-предшественником, содержание которого в фибрилле достигает 80% и является специфичным признаком для каждого типа амилоидоза.
Характеристика основных типов амилоидоза
В группу АА-амилоидоза входят реактивный (вторичный) амилоидоз, амилоидоз при периодической болезни, криопиринопатиях (преимущественно при синдроме Макла – Уэллса – семейной периодической лихорадке в сочетании с глухотой и крапивницей). Наиболее частыми причинами вторичного амилоидоза в настоящее время служат ревматоидный артрит (30–50%), хронические гнойно-деструктивные болезни (остеомиелит, бронхоэктатическая болезнь), воспалительные заболевания кишечника (язвенный колит, болезнь Крона), туберкулез, опухоли (чаще лимфогранулематоз и рак почки) [3].
Наиболее частыми причинами вторичного амилоидоза в настоящее время служат ревматоидный артрит (30–50%), хронические гнойно-деструктивные болезни (остеомиелит, бронхоэктатическая болезнь), воспалительные заболевания кишечника (язвенный колит, болезнь Крона), туберкулез, опухоли (чаще лимфогранулематоз и рак почки) [3].
АА-амилоид образуется из сывороточного белка-предшественника SAA – острофазового белка, в норме синтезируемого гепатоцитами, нейтрофилами и фибробластами в следовых количествах. Его концентрация значительно возрастает под воздействием интерлейкинов 1 и 6, фактора некроза опухоли в ответ на воспаление, опухолевый рост. Повышение содержания SAA в крови играет основную роль в патогенезе АА-амилоидоза.
Однако для развития амилоидоза недостаточно только высокой концентрации SAA, необходимо также наличие у белка-предшественника амилоидогенности. Развитие амилоидоза у человека связывают с депозицией SAA1; в настоящее время известно 5 изотипов SAA1, из которых наибольшую амилоидогенность приписывают изотипам 1. 1 и 1.5 [4]. Конечный этап амилоидогенеза – образование фибрилл амилоида из белка-предшественника – осуществляется при неполном расщеплении протеазами моноцитов-макрофагов. Стабилизация амилоидной фибриллы и резкое снижение растворимости этого макромолекулярного комплекса во многом обусловлены взаимодействием с полисахаридами интерстиция. При АА-амилоидозе амилоид обнаруживают в различных органах, однако клиническая картина и прогноз обусловлены поражением почек.
1 и 1.5 [4]. Конечный этап амилоидогенеза – образование фибрилл амилоида из белка-предшественника – осуществляется при неполном расщеплении протеазами моноцитов-макрофагов. Стабилизация амилоидной фибриллы и резкое снижение растворимости этого макромолекулярного комплекса во многом обусловлены взаимодействием с полисахаридами интерстиция. При АА-амилоидозе амилоид обнаруживают в различных органах, однако клиническая картина и прогноз обусловлены поражением почек.
AL-амилоидоз включает в себя первичный (идиопатический) амилоидоз и амилоидоз, ассоциированный с миеломной болезнью, развивающийся у 7–10% больных с множественной миеломой. По современным представлениям, первичный AL-амилоидоз и миеломную болезнь (как ассоциированную, так и не ассоциированную с амилоидозом) рассматривают в рамках единой В-лимфоцитарной дискразии, которая характеризуется пролиферацией аномального клона плазматических клеток или В-клеток в костном мозге с избыточной продукцией моноклонального иммуноглобулина, обладающего амилоидогенностью. Белком-предшественником при AL-амилоидозе считают моноклональные легкие цепи иммуноглобулинов (ЛЦИ), из названия которых происходит аббревиатура L [3]. AL-амилоидоз – это генерализованный процесс с преимущественным поражением сердца, почек, желудочно-кишечного тракта (ЖКТ), нервной системы и кожи.
Белком-предшественником при AL-амилоидозе считают моноклональные легкие цепи иммуноглобулинов (ЛЦИ), из названия которых происходит аббревиатура L [3]. AL-амилоидоз – это генерализованный процесс с преимущественным поражением сердца, почек, желудочно-кишечного тракта (ЖКТ), нервной системы и кожи.
К ATTR-амилоидозу относят семейную амилоидную полиневропатию, наследуемую по аутосомно-доминантному типу, и системный старческий амилоидоз. Белком-предшественником при этой форме амилоидоза служит транстиретин – компонент фракции преальбумина, синтезируемый печенью и выполняющий функции транспортного белка тироксина и ретинола. Установлено, что АTTR-амилоидоз бывает результатом мутации в гене, кодирующем синтез транстиретина, что приводит к замене аминокислот в молекуле TTR. В результате повышается амилоидогенность белка-предшественника и облегчается его полимеризация в амилоидные фибриллы. В настоящее время известно множество вариантных транстиретинов, чем и обусловлено разнообразие клинических форм наследственной невропатии [5]. Эта группа заболеваний характеризуется прогрессирующей периферической и вегетативной невропатией, которая сочетается с поражением сердца, почек и других органов различной степени.
Эта группа заболеваний характеризуется прогрессирующей периферической и вегетативной невропатией, которая сочетается с поражением сердца, почек и других органов различной степени.
Системный старческий амилоидоз развивается после 70 лет в результате возрастных изменений структуры нормального транстиретина, усиливающих его амилоидогенность. К органам-мишеням старческого амилоидоза относят сердце, сосуды головного мозга и аорту.
К семейным формам амилоидоза относятся также более редкие AGel-, AFib-, ALys-амилоидозы, при которых амилоидогенностью обладают мутантные формы соответственно гелсолина, фибриногена, лизоцима. При этих вариантах амилоидоза также отмечается преимущественное поражение почек, однако для гелсолинового амилоидоза характерно сочетание нефропатии с сетчатой дистрофией роговицы и периферической невропатией (преимущественно поражаются черепно-мозговые нервы).
В настоящее время известно более 20 амилоидогенных белков-предшественников и, соответственно, клинических вариантов амилоидоза. Так, А-бета-амилоид является морфологической основой болезни Альцгеймера, AIAPP-амилоид – сахарного диабета 2 типа и др. [3].
Так, А-бета-амилоид является морфологической основой болезни Альцгеймера, AIAPP-амилоид – сахарного диабета 2 типа и др. [3].
В нефрологической практике большое значение имеет А-бета-2-М-амилоидоз (ассоциированный с хроническим гемодиализом). Белком-предшественником при этой форме амилоидоза служит бета-2-микроглобулин, который в норме присутствует в крови, моче, спинномозговой и синовиальной жидкостях. При нормальной функции почек его концентрация в крови составляет 1–2 мг/л. Этот белок фильтруется в клубочках почек и метаболизируется после реабсорбции в проксимальных канальцах. У пациентов с ХПН концентрация этого белка в крови возрастает, коррелируя с содержанием креатинина, однако максимальных значений (в 20–70 раз превышающих норму) она достигает через несколько лет проведения регулярного гемодиализа. Бета-2-микроглобулин плохо удаляется при проведении стандартного гемодиализа, что неизбежно приводит к развитию амилоидоза [3]. Первые депозиты бета-2-М-амилоида обнаруживают в синовиальной оболочке суставов уже через 1–2 года заместительной почечной терапии, клинические симптомы появляются через 7 и более лет терапии [3]. У больных старше 60 лет диализный амилоидоз развивается быстрее. Кроме высокой концентрации белка-предшественника, в патогенезе диализного амилоидоза существенную роль играют и другие факторы. Амилоидогенность бета-2-микроглобулина возрастает при модификациях, обусловленных взаимодействием с продуктами неполного гликирования, а также при окислении бета-2-микроглобулина и ацидификации среды [3]. Одним из основных источников этих модификаций являются компоненты диализата, в том числе бактериальный липополисахарид; гликирование бета-2-микроглобулина возможно также при контакте с целлюлозными диализными мембранами. Таким образом, профилактика диализного амилоидоза во многом связана с отказом от целлюлозных диализных мембран и тщательностью контроля качества применяемого диализата. Модифицированный бета-2-микроглобулин является мощным индуктором образования цитокинов (интерлейкины 1 и 6, фактор некроза опухоли альфа). Результатом активации цитокинового каскада является тяжелое синовиальное воспаление с привлечением макрофагов [3].
У больных старше 60 лет диализный амилоидоз развивается быстрее. Кроме высокой концентрации белка-предшественника, в патогенезе диализного амилоидоза существенную роль играют и другие факторы. Амилоидогенность бета-2-микроглобулина возрастает при модификациях, обусловленных взаимодействием с продуктами неполного гликирования, а также при окислении бета-2-микроглобулина и ацидификации среды [3]. Одним из основных источников этих модификаций являются компоненты диализата, в том числе бактериальный липополисахарид; гликирование бета-2-микроглобулина возможно также при контакте с целлюлозными диализными мембранами. Таким образом, профилактика диализного амилоидоза во многом связана с отказом от целлюлозных диализных мембран и тщательностью контроля качества применяемого диализата. Модифицированный бета-2-микроглобулин является мощным индуктором образования цитокинов (интерлейкины 1 и 6, фактор некроза опухоли альфа). Результатом активации цитокинового каскада является тяжелое синовиальное воспаление с привлечением макрофагов [3]. Макрофагальное воспаление определяет высокий деструктивный потенциал воспаления в синовии с развитием костных эрозий и кист, рост которых обусловливает развитие переломов костей. Было установлено, что бета-2-микроглобулин обладает высокой коллагенсвязывающей активностью, возрастающей по мере увеличения его концентрации в крови. Кроме того, выявлена связь бета-2-микроглобулина с гликозоаминогликанами хряща. Этими фактами можно объяснить преимущественное отложение фибрилл амилоида в суставных тканях. Таким образом, при А-бета-2-М-амилоидозе отмечают поражение костей и периартикулярных тканей, реже – сосудов.
Макрофагальное воспаление определяет высокий деструктивный потенциал воспаления в синовии с развитием костных эрозий и кист, рост которых обусловливает развитие переломов костей. Было установлено, что бета-2-микроглобулин обладает высокой коллагенсвязывающей активностью, возрастающей по мере увеличения его концентрации в крови. Кроме того, выявлена связь бета-2-микроглобулина с гликозоаминогликанами хряща. Этими фактами можно объяснить преимущественное отложение фибрилл амилоида в суставных тканях. Таким образом, при А-бета-2-М-амилоидозе отмечают поражение костей и периартикулярных тканей, реже – сосудов.
Патогенез амилоидоза
Несмотря на различие в типах амилоидного белка, существует общность патогенеза различных клинических форм амилоидоза. Основной причиной развития болезни служит наличие определенного, нередко повышенного количества амилоидогенного предшественника. Как уже указывалось, появление или усиление амилоидогенности может быть обусловлено циркуляцией вариантов белков с повышенной общей гидрофобностью молекулы, нарушенным соотношением поверхностных молекулярных зарядов, что приводит к нестабильности белковой молекулы и способствует ее агрегации в амилоидную фибриллу. Эти механизмы особенно ярко прослеживаются на примере белков, в функцию которых заложена необходимость физиологического изменения конформации. Так, практически все аполипопротеины, вынужденные разворачивать свою вторичную структуру в процессе транслокации холестерина через стенку сосуда, участвуют в патогенезе различных форм амилоидоза. На последнем этапе амилоидогенеза происходит взаимодействие амилоидного белка с белками плазмы крови и гликозоаминогликанами тканей. Кроме структурных особенностей, имеют значение также физико-химические свойства межклеточного матрикса, где происходит сборка амилоидной фибриллы. В практике экспериментального амилоидоза хорошо известна способность суспензии амилоидных масс (амилоидускоряющая субстанция), полученной из тканей животных, пораженных амилоидом, провоцировать амилоидоз при введении здоровым животным. В клинической практике у больных ATTR-амилоидозом, несмотря на прекращение циркуляции патологического транстиретина после трансплантации здоровой печени, продолжается нарастание массы амилоидных депозитов в сердце за счет захвата нормального неизмененного транстиретина [6].
Эти механизмы особенно ярко прослеживаются на примере белков, в функцию которых заложена необходимость физиологического изменения конформации. Так, практически все аполипопротеины, вынужденные разворачивать свою вторичную структуру в процессе транслокации холестерина через стенку сосуда, участвуют в патогенезе различных форм амилоидоза. На последнем этапе амилоидогенеза происходит взаимодействие амилоидного белка с белками плазмы крови и гликозоаминогликанами тканей. Кроме структурных особенностей, имеют значение также физико-химические свойства межклеточного матрикса, где происходит сборка амилоидной фибриллы. В практике экспериментального амилоидоза хорошо известна способность суспензии амилоидных масс (амилоидускоряющая субстанция), полученной из тканей животных, пораженных амилоидом, провоцировать амилоидоз при введении здоровым животным. В клинической практике у больных ATTR-амилоидозом, несмотря на прекращение циркуляции патологического транстиретина после трансплантации здоровой печени, продолжается нарастание массы амилоидных депозитов в сердце за счет захвата нормального неизмененного транстиретина [6]. Многие формы амилоидоза можно объединить также по признаку возникновения в пожилом и старческом возрасте (AL, ATTR, AIAPP, AApoA1, AFib, ALys, AANF, A-бета), что указывает на наличие механизмов возрастной эволюции структуры определенных белков в сторону повышения амилоидогенности и позволяет рассматривать амилоидоз как одну из моделей старения организма.
Многие формы амилоидоза можно объединить также по признаку возникновения в пожилом и старческом возрасте (AL, ATTR, AIAPP, AApoA1, AFib, ALys, AANF, A-бета), что указывает на наличие механизмов возрастной эволюции структуры определенных белков в сторону повышения амилоидогенности и позволяет рассматривать амилоидоз как одну из моделей старения организма.
Классификация амилоидоза
Объяснение многообразия клинических форм амилоидоза наличием многочисленных сывороточных белков-предшественников привело к созданию современной классификации амилоидоза, в основу которой положен биохимический тип белка-предшественника (табл.). Все типы амилоидоза обозначают аббревиатурой, в которой первая буква А означает «амилоидоз», а последующие – сокращенное название основных фибриллярных белков амилоида: А – амилоидный белок А, L – легкие цепи иммуноглобулинов, TTR – транстиретин, бета-2-М – бета2-микроглобулин и др. С клинической точки зрения целесообразно выделять локальные и системные, или генерализованные, формы амилоидоза. Среди последних основными считают АА-, AL-, АTTR- и А-бета-2-М-амилоидоз.
Среди последних основными считают АА-, AL-, АTTR- и А-бета-2-М-амилоидоз.
Симптомы амилоидоза почек
В клинической практике наибольшее значение имеют АА- и AL-типы системного амилоидоза, которые протекают с вовлечением в патологический процесс многих органов, однако чаще манифестируют симптомами моноорганного поражения. В дальнейшем развивается, как правило, столь характерный для этих типов амилоидоза полиморфизм клинических проявлений. АА- и AL-амилоидоз у мужчин отмечают в 1,8 раза чаще, чем у женщин. Для вторичного амилоидоза характерно более раннее начало, чем для первичного (средний возраст заболевших – около 40 и 65 лет соответственно). Среди многочисленных клинических проявлений АА- и AL-амилоидоза наряду с общими для обоих типов симптомами существуют характерные лишь для AL-типа признаки (периорбитальные геморрагии, макроглоссия, поражение кожи и др.) [7–9]. Кроме того, клинические проявления, напоминающие признаки первичного амилоидоза, возможны при АTTR- и А-бета-2-М-амилоидозе (синдром запястного канала и др. ).
).
Поражение почек – ведущий клинический признак АА- и AL-амилоидоза. При АА-амилоидозе почки вовлечены в патологический процесс практически у всех больных, при AL-типе частота нефропатии также высока и приближается к 80%. Поражение почек наблюдают и при АTTR-амилоидозе, однако у большинства больных семейной амилоидной невропатией отмечают несоответствие между клиническими и морфологическими признаками нарушений почек.
Амилоид при АА- и AL-типах амилоидоза локализуется преимущественно в клубочках, однако у 10% больных первичным амилоидозом и у значительной части больных наследственной невропатией отмечают только отложения вне клубочков. Амилоид откладывается также в других почечных структурах: в базальной мембране канальцев (преимущественно дистальных и петли Генле), интерстиции, стенках сосудов.
Клинически амилоидная нефропатия манифестирует, как правило, изолированно протеинурией и характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурическая, нефротическая, ХПН.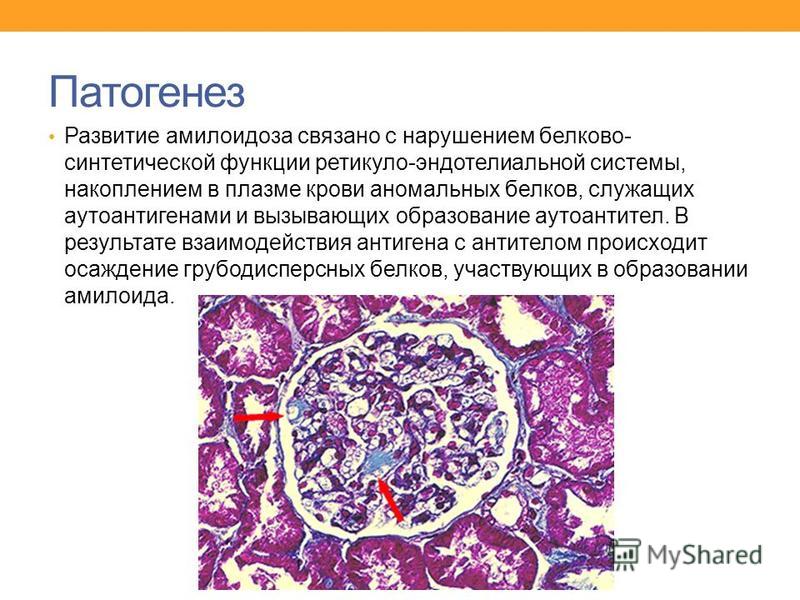 Только у 20% больных АА-амилоидозом ХПН развивается без предшествующего нефротического синдрома. При AL-амилоидозе стадийность течения амилоидной нефропатии проявляется менее отчетливо [9].
Только у 20% больных АА-амилоидозом ХПН развивается без предшествующего нефротического синдрома. При AL-амилоидозе стадийность течения амилоидной нефропатии проявляется менее отчетливо [9].
К особенностям амилоидоза почек относят редкость гематурии и лейкоцитурии («скудный» мочевой осадок), а также артериальной гипертонии, которую даже при ХПН отмечают лишь у 20% больных АА-амилоидозом и еще реже при AL-амилоидозе. Нефротический синдром и большие размеры почек сохраняются даже при развитии и прогрессировании ХПН.
Величина протеинурии не коррелирует с выраженностью амилоидных отложений в почках (при преимущественно сосудистом поражении протеинурия может быть минимальной) и зависит от степени деструкции подоцитов. Максимальную потерю белка обнаруживают через участки базальной мембраны, которые пропитаны амилоидом и лишены эпителиального покрытия.
Амилоидоз почек у большинства больных диагностируют на стадии нефротического синдрома, у 33% – на стадии ХПН. В редких случаях амилоидная нефропатия может проявляться остронефротическим синдромом и макрогематурией. Описаны также синдром Фанкони и тромбоз почечных вен.
Описаны также синдром Фанкони и тромбоз почечных вен.
Поражение сердца отмечают у подавляющего большинства больных AL-амилоидозом и у части пациентов с АTTR-амилоидозом, в то время как для АА-амилоидоза этот симптом не характерен. В результате замещения миокарда амилоидными массами развивается рестриктивная кардиомиопатия [10]. Клинически определяют кардиомегалию, рано развивается сердечная недостаточность (у 22% больных уже в дебюте болезни), которая быстро прогрессирует и почти у 50% пациентов, наряду с аритмиями, бывает причиной смерти. Особенностью сердечной недостаточности при первичном амилоидозе служит ее рефрактерность к терапии, в первую очередь сердечными гликозидами [11].
Нарушения ритма и проводимости при AL-амилоидозе многообразны: мерцательная аритмия, наджелудочковая тахикардия, синдром преждевременного возбуждения желудочков, различные блокады и синдром слабости синусового узла. Вследствие отложения амилоида в коронарных артериях возможно развитие инфаркта миокарда, обнаруживаемого на аутопсии у 6% больных. Амилоидные отложения в клапанных структурах симулируют картину клапанного порока. Основным признаком амилоидоза сердца на электрокардиограмме бывает снижение вольтажа зубцов комплекса QRS. Описан инфарктоподобный тип электрокардиограммы. Наиболее адекватным методом диагностики амилоидной кардиомиопатии считают эхокардиографию, с помощью которой можно диагностировать симметричное утолщение стенок желудочков, дилатацию предсердий, нарушения гемодинамики.
Серьезным патологическим признаком при AL-амилоидозе служит ортостатическая артериальная гипотония, которую наблюдают у 11% больных в момент постановки диагноза. Обычно этот симптом связан с автономной дисфункцией (поражение вегетативной нервной системы) и в тяжелых случаях сопровождается синкопальными состояниями. Артериальная гипотония бывает также у больных АА-амилоидозом, но в этом случае ее причиной служит надпочечниковая недостаточность вследствие отложения амилоида в надпочечниках [3].
Поражение дыхательной системы возникает при первичном амилоидозе примерно у 50% больных, а при вторичном – у 10–14%. В большинстве случаев оно протекает бессимптомно или со скудной клинической симптоматикой [12]. При AL-амилоидозе одним из ранних признаков болезни может быть охриплость или изменение тембра голоса вследствие отложения амилоида в голосовых связках, опережающего его появление в дистальных отделах дыхательных путей. В легких амилоид откладывается преимущественно в альвеолярных перегородках (что приводит к развитию кашля и одышки) и стенках сосудов. Описаны также ателектазы и инфильтраты в легких. Рентгенологическая картина не специфична, смерть от прогрессирующей дыхательной недостаточности наступает редко [13].
Поражение органов пищеварения наблюдают при амилоидозе в 70% случаев, причем у больных с AL- и АА-типами амилоидоза частота поражения тех или иных отделов ЖКТ различна [3]. У 25% больных первичным амилоидозом отмечают амилоидное поражение пищевода, проявляющееся преимущественно дисфагией, которая может быть одним из ранних симптомов заболевания. О поражении желудка и кишечника могут свидетельствовать изъязвления и перфорации их стенок с возможным кровотечением, а также препилорическая непроходимость желудка или механическая кишечная непроходимость из-за отложения амилоидных масс. У больных с преимущественным поражением толстой кишки возможно появление клинических симптомов, имитирующих язвенный колит. Наиболее частым желудочно-кишечным проявлением AL-амилоидоза, отмечаемым почти у 25% пациентов, бывает тяжелая моторная диарея с вторичным нарушением всасывания. Возможной причиной тяжелой диареи при этом, наряду с инфильтрацией кишечной стенки, и в том числе ворсин, амилоидом, у больных AL-амилоидозом служит автономная (вегетативная) дисфункция. Истинный синдром нарушенного всасывания развивается приблизительно у 4–5% больных. При АА-амилоидозе эти симптомы иногда также возможны, в том числе как единственное клиническое проявление амилоидоза.
Поражение печени при АА- и AL-амилоидозе наблюдают практически в 100% случаев, при этом обычно отмечают небольшое увеличение печени и 3–4-кратное повышение активности гамма-глутамилтранспептидазы и щелочной фосфатазы [14]. Увеличению печени обычно сопутствует небольшое увеличение селезенки. Тяжелое поражение печени с выраженной гепатомегалией и развернутыми признаками тяжелого холестаза отмечается значительно реже (у 15–25% больных) и более характерно для AL-амилоидоза. Несмотря на выраженную гепатомегалию, функция печени обычно остается сохранной. Редким признаком амилоидоза печени бывает внутрипеченочная портальная гипертензия, которая чаще сочетается с выраженной желтухой, холестазом, печеночной недостаточностью и свидетельствует о далеко зашедшем поражении печени с риском пищеводного кровотечения, печеночной комы. При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения [15]. Редким проявлением амилоидоза селезенки бывает ее спонтанный разрыв.
Поражение нервной системы, представленное симптомами периферической невропатии и вегетативной дисфункции, отмечают у 17% больных AL-амилоидозом и у пациентов с семейной амилоидной невропатией разных типов (ATTR, AApoA1 и др.). Клиническая картина невропатии при всех типах амилоидоза практически одинакова, поскольку обусловлена сходными процессами: в первую очередь, дегенерацией миелиновой оболочки нервов, а также компрессией нервных стволов отложениями амилоида и ишемией в результате амилоидных депозитов в стенках сосудов [3].
В большинстве случаев возникает симметричная дистальная невропатия с неуклонным прогрессированием. В дебюте поражения нервной системы наблюдают, главным образом, сенсорные нарушения, в первую очередь болевой и температурной чувствительности, позже присоединяются нарушения вибрационной и позиционной чувствительности, двигательные нарушения. Ранними симптомами невропатии бывают парестезии или мучительные дизестезии (чувство онемения). Нижние конечности вовлекаются в патологический процесс чаще, чем верхние.
Автономные дисфункции часто манифестируют ортостатической артериальной гипотонией (см. выше), иногда с обморочными состояниями, диареей, нарушением функции мочевого пузыря, импотенцией у мужчин.
У 20% больных AL-амилоидозом и у большинства пациентов с амилоидозом на фоне проведения гемодиализа выявляют синдром запястного канала, обусловленный сдавлением срединного нерва в запястном канале амилоидом, откладывающимся в связках запястья [2, 8, 9]. Клинически этот синдром проявляется интенсивными болями и парестезиями в I–III пальцах кисти с постепенной атрофией мышц тенара. К особенностям синдрома запястного канала при диализном амилоидозе относят его преимущественное развитие на той руке, где сформирована фистула, а также усиление болей во время процедуры гемодиализа, возможно, в результате развития феномена обкрадывания, индуцированного фистулой, что приводит к ишемии срединного нерва.
Поражение кожи наблюдают почти у 40% больных первичным амилоидозом. Для него характерно разнообразие проявлений, наиболее частыми из которых бывают параорбитальные геморрагии (патогномоничны для AL-амилоидоза), возникающие при малейшем напряжении. Описаны также папулы, бляшки, узелки, пузырьковые высыпания. Нередко наблюдают индурацию кожи, аналогичную склеродермической. Редким вариантом поражения кожи при AL-амилоидозе служат нарушения пигментации (от выраженного усиления до тотального альбинизма), алопеция, трофические нарушения.
Поражение опорно-двигательного аппарата характерно для пациентов с диализным амилоидозом и редко (в 5–10% случаев) возникает у больных AL-амилоидозом (исключая костные изменения при миеломной болезни). При этом характер тканевого отложения амилоида сходен при обоих этих типах амилоидоза: амилоид откладывается в костях, суставном хряще, синовии, связках и мышцах [16, 17].
При диализном амилоидозе наиболее часто отмечают триаду признаков – плече-лопаточный периартрит, синдром запястного канала и поражение сухожильных влагалищ сгибателей кисти, приводящее к развитию сгибательных контрактур пальцев. Кроме них, характерно развитие кистозного поражения костей из-за отложения амилоида. Типичны амилоидные кисты в костях запястья и головках трубчатых костей. Со временем эти отложения увеличиваются в размерах, становясь причиной патологических переломов.
Частым признаком диализного амилоидоза бывает также деструктивная спондилоартропатия в результате амилоидного поражения межпозвонковых дисков, преимущественно в шейном отделе позвоночника.
Амилоидные отложения в мышцах чаще наблюдают при первичном амилоидозе. Они проявляются псевдогипертрофией или атрофией мышц, затрудняющими движения, мышечными болями. Макроглоссия, обусловленная выраженной инфильтрацией мышц амилоидом, – это патогномоничный симптом AL-амилоидоза, который встречается примерно у 20% пациентов и нередко сочетается с псевдогипертрофией других групп поперечно-полосатой мускулатуры. В тяжелых случаях макроглоссия не только затрудняет прием пищи и речь, но и приводит к обструкции дыхательных путей [3]. При АА-амилоидозе она не развивается.
Среди других органных поражений при амилоидозе известны поражение щитовидной железы с развитием клинической картины гипотиреоза (AL-амилоидоз), надпочечников с появлением симптомов их недостаточности (чаще при АА-амилоидозе), экзокринных желез, приводящее к возникновению сухого синдрома, лимфаденопатия. Редким проявлением, описанным при AL- и АTTR- амилоидозе, бывает поражение глаз.
Диагностика амилоидоза
Предполагаемый на основании клинических и лабораторных данных амилоидоз необходимо подтвердить морфологически обнаружением амилоида в биоптатах тканей.
При подозрении на AL-тип амилоидоза рекомендуют производить пункцию костного мозга. Подсчет плазматических клеток и окраска пунктата на амилоид позволяют не только диагностировать амилоидоз, но и дифференцировать первичный и ассоциированный с миеломой варианты AL-амилоидоза. Положительный результат исследования костного мозга на амилоид отмечают у 60% больных AL-амилоидозом.
Простой и безопасной диагностической процедурой считают аспирационную биопсию подкожной жировой клетчатки, при которой выявляют амилоид в 80% случаев AL-амилоидоза. К преимуществам этой процедуры, кроме информативности, относят также редкость развития кровотечений, что позволяет использовать этот метод у больных с нарушениями свертывания крови (больные первичным амилоидозом нередко имеют дефицит Х фактора свертывания, при котором могут развиться геморрагии [8]).
Наиболее часто для диагностики разных типов амилоидоза проводят биопсию слизистой оболочки прямой кишки, почки, печени. Биопсия слизистого и подслизистого слоев прямой кишки позволяет выявить амилоид у 70% больных, а биопсия почки – практически в 100% случаев. У пациентов с синдромом запястного канала исследованию на амилоид необходимо подвергать ткань, удаленную при операции декомпрессии запястного канала.
Биопсийный материал для выявления амилоида необходимо окрашивать конго красным с последующей микроскопией в поляризованном свете для выявления способности к двойному лучепреломлению. Современная морфологическая диагностика амилоидоза включает не только обнаружение, но и типирование амилоида, поскольку тип амилоида определяет терапевтическую тактику. Для типирования часто применяют пробу с перманганатом калия. При обработке окрашенных конго красным препаратов 5%-ным раствором перманганата калия АА-тип амилоида теряет окраску и утрачивает свойство двойного лучепреломления, тогда как AL-тип амилоида сохраняет их. Использование щелочного гуанидина позволяет более точно дифференцировать АА- и AL-амилоидоз.
Наиболее эффективным методом типирования амилоида служит иммуногистохимическое исследование с применением антисывороток к основным типам амилоидного белка (специфические антитела против АА-белка, легких цепей иммуноглобулинов, транстиретина и бета-2-микроглобулина) [3, 18].
Лечение амилоидоза
Согласно современным представлениям, целью терапии любого типа амилоидоза является уменьшение количества (или, если возможно, удаление) белков-предшественников для того, чтобы замедлить или приостановить прогрессирование болезни. Неблагоприятный прогноз при естественном течении амилоидоза оправдывает применение некоторых агрессивных лекарственных режимов или других радикальных мер. Клиническое улучшение, достигаемое с помощью этих видов лечения, включает стабилизацию или восстановление функции жизненно важных органов, а также предотвращение функциональных нарушений, что увеличивает продолжительность жизни больных. Морфологическим критерием эффективности лечения считают уменьшение отложений амилоида в тканях, что в настоящее время можно оценить, применяя радиоизотопную сцинтиграфию с сывороточным Р-компонентом амилоида. Кроме основных терапевтических режимов, лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности застойной недостаточности кровообращения, аритмии, отечного синдрома, коррекцию артериальной гипер- и гипотонии.
Целью терапии вторичного амилоидоза служит подавление продукции белка-предшественника SAA [19, 20], что достигается лечением хронического воспаления, в том числе и хирургическим (секвестрэктомия при остеомиелите, удаление доли легкого при бронхоэктатической болезни, опухоли, туберкулезе).
Особое значение в настоящее время придают терапии ревматоидного артрита, учитывая его лидирующее положение среди причин вторичного амилоидоза. Базисная терапия ревматоидного артрита цитостатическими лекарственными средствами (метотрексатом, циклофосфамидом, хлорамбуцилом) или современными антицитокиновыми средствами (блокаторы эффектов фактора некроза опухоли альфа, интерлейкины 1 и 6, блокаторы СD20 В-лимфоцитов и др.), назначаемая на длительный срок (более 12 мес.), уменьшает риск развития амилоидоза. У пациентов с уже развившимся амилоидозом это лечение позволяет в большинстве случаев уменьшить клинические проявления амилоидной нефропатии. В результате подобной терапии отмечают снижение выраженности протеинурии, купирование нефротического синдрома, стабилизацию функции почек. У части пациентов удается предотвратить развитие ХПН или замедлить ее прогрессирование, что существенно улучшает прогноз. Об эффективности лечения свидетельствует также нормализация концентрации С-реактивного белка в крови.
Средством выбора для лечения АА-амилоидоза при периодической болезни служит колхицин. При его постоянном приеме можно полностью избежать рецидивирования приступов у большинства больных, предотвращая у них также развитие амилоидоза. При развившемся амилоидозе длительный, возможно пожизненный, прием колхицина в дозе 1,8–2 мг/сут приводит к ремиссии, выражающейся в ликвидации нефротического синдрома, уменьшении или исчезновении протеинурии у больных с нормальной функцией почек. При наличии ХПН начальную дозу лекарственных средств уменьшают в зависимости от величины клубочковой фильтрации, хотя в случае снижения концентрации креатинина в крови возможно повышение дозы до стандартной. Колхицин также предотвращает рецидив амилоидоза в трансплантированной почке. Больные хорошо переносят данное лекарственное средство. Если эффективность колхицина при амилоидозе в рамках периодической болезни не вызывает сомнений, то имеются лишь единичные работы, свидетельствующие о его успешном применении у больных вторичным амилоидозом. Кроме колхицина, при АА-амилоидозе применяют диметилсульфоксид, вызывающий резорбцию амилоидных отложений. Однако использование его в высоких дозах (не менее 10 г/сут), необходимых для успешного лечения, ограничено из-за крайне неприятного запаха.
При AL-амилоидозе, как и при миеломной болезни, лечение должно быть направлено на подавление пролиферации клона плазматических клеток для уменьшения продукции ЛЦИ. Наибольший опыт лечения накоплен для мелфалана в сочетании с преднизолоном [21]. Лечение продолжают 7-дневными курсами с интервалом 4–6 недель на протяжении 12–24 месяцев. Доза мелфалана составляет 0,15 мг/кг массы тела в сутки, преднизолона – 0,8 мг/кг массы тела в сутки. У больных с ХПН (скорость клубочковой фильтрации менее 40 мл/мин) дозу мелфалана уменьшают на 50%. Эта схема лечения характеризуется низким риском осложнений химиотерапии и может применяться у абсолютного большинства больных, даже при наличии тяжелых висцеральных проявлений амилоидоза. Однако эффект этой терапии достигается только у 18–30% больных.
В настоящее время разработан эффективный иммунохимический метод оценки ремиссии плазмоклеточной дискразии у больных амилоидозом – определение свободных ЛЦИ в сыворотке (Freelite). Ранее применявшиеся методы не позволяли отличить пул свободных ЛЦИ от пула связанных с иммуноглобулинами, что резко снижало чувствительность методов. Благодаря методу Freelite, который базируется на применении антител к скрытым эпитопам ЛЦИ, появилась возможность количественной оценки пула свободных ЛЦИ, впервые были сформулированы критерии нормальной продукции ЛЦИ. В настоящее время во всем мире метод Freelite стал стандартом оценки эффективности терапии и мониторирования течения болезни у больных амилоидозом [22, 23].
С целью повышения эффективности лечения широко применялись попытки лечения более агрессивными схемами химиотерапии с включением винкристина, доксорубицина, циклофосфана, мелфалана, дексаметазона в разных комбинациях. Наибольшая эффективность достигнута при химиотерапии с использованием высоких доз. Так, в 1996 г. были опубликованы предварительные результаты (R.L. Comenzo и соавт.) лечения 5 больных AL-амилоидозом внутривенными вливаниями мелфалана в дозе 200 мг/м² поверхности тела с последующим введением аутологичных стволовых клеток (CD34+) крови [24]. Аутологичные стволовые клетки получают методом лейкофереза крови больного после предварительной их мобилизации из костного мозга под влиянием введенного извне гранулоцитарного колониестимулирующего фактора. Однако тяжелый агранулоцитоз и другие осложнения этой терапии существенно ограничивают применение терапии сверхвысокими дозами мелфалана, в частности, у больных с недостаточностью кровообращения. В последние годы показана высокая эффективность при относительно удовлетворительной переносимости схемы «мелфалан (0,15 мг/кг 4 дня) в сочетании с дексаметазоном (20 мг/сут в 1–4-е и 9–12-е дни)» ежемесячными курсами [25]. В настоящее время эта схема стала первой линией терапии AL-амилоидоза. В качестве второй линии терапии обсуждаются схемы с включением бортезомиба и талидомида [26, 27]. Кардиотоксичность и нейротоксичность этих препаратов не позволяют рекомендовать их в качестве препаратов первой линии выбора, несмотря на высокую эффективность в отношении плазмоклеточной дискразии.
Основной целью лечения А-бета-2-М-амилоидоза является уменьшение поступления в ткани бета-2-микроглобулина. Поскольку способы подавления его продукции в настоящее время отсутствуют, необходимо увеличить клиренс бета-2-микроглобулина путем проведения современных методов очищения крови.
Широко распространено мнение о том, что наиболее эффективным методом выведения бета-2-микроглобулина является трансплантация почки. Эта точка зрения подкреплена высокой эффективностью трансплантации в отношении костных болей, характерных для диализного амилоидоза. В настоящее время установлено, что причиной костных болей, так же как и костных кист, является макрофагальное воспаление, характерное для этого типа амилоидоза. Быстрое исчезновение болей после трансплантации является следствием назначения стероидов и цитостатиков, необходимых для предотвращения отторжения трансплантата и одновременно подавляющих макрофагальное воспаление в зоне депозиции амилоида. На поздних этапах амилоидогенеза зависимость макрофагального воспаления от поставки бета-2-микроглобулина в ткани снижается, и улучшение клиренса этого белка после трансплантации не предупреждает дальнейшее прогрессирование костных кист и эрозий. Таким образом, трансплантация способна предотвратить прогрессирование диализного амилоидоза только на ранних, в основном доклинических, стадиях амилоидоза [28].
В этой связи в лечении диализного амилоидоза большое значение придают современным диализным технологиям увеличения клиренса бета-2-микроглобулина. При проведении стандартной процедуры гемодиализа с использованием низкопоточных мембран (диффузионная модель) клиренс бета-2-микроглобулина существенно ограничен, так как этот белок по своей массе (11 800 Да) приближается к классу так называемых средних молекул. Однако применение высокопоточных синтетических мембран и сочетание диффузионного механизма очищения крови с конвективным приводят к существенному повышению эффективности выведения бета-2-микроглобулина. Так, было показано, что лечение гемодиафильтрацией в режиме online (режим ГДФ ONLINE предусматривает использование в качестве замещающего раствора сверхчистого диализата, который готовится непосредственно в аппарате для гемодиализа) позволяет добиться значительного снижения постдиализного уровня бета-2-микроглобулина (на 70% и более) [29]. В этой связи наиболее предпочтительным методом гемодиализа с точки зрения профилактики и лечения диализного амилоидоза является высокоэффективная ГДФ ONLINE с использованием синтетических мембран и скоростью подачи замещающего раствора не менее 100 мл/мин. Пациенты, которые получали ГДФ ONLINE с объемом замещения не менее 24 л за 4 часа процедуры, имели достоверно более существенное (на 72,7%) снижение сывороточного уровня бета-2-микроглобулина по сравнению с пациентами, получавшими стандартный гемодиализ на низкопоточных мембранах (на 49,7%) [30].
Результаты недавно завершенного исследования CONTRAST (CONvective TRAnsport STudy), посвященного влиянию конвективного транспорта на сывороточный уровень бета-2-микроглобулина, свидетельствуют о том, что через 6 месяцев у пациентов, которым проводится гемодиафильтрация, уровень бета-2-микроглобулина в сыворотке достоверно ниже, чем у пациентов на низкопоточном гемодиализе [31].
Дальнейшие перспективы в развитии гемодиализа во многом связаны с технологическим усовершенствованием и широким распространением методики ГДФ ONLINE в клинической практике. В частности, усовершенствованная техника ГДФ ONLINE – MIXED ГДФ – позволяет существенно увеличить эффективность конвективного транспорта, в том числе перенос средних молекул, в безопасном для пациента режиме [32–34].
Большое значение имеет применение современных синтетических диализных мембран, которые способны адсорбировать значительные количества бета-2-микроглобулина, в результате чего концентрация этого белка в крови в течение процедуры гемодиализа может снижаться на 30–40% [29]. Разработаны также специальные сорбционные колонки с высокими показателями очищения крови от бета-2-микроглобулина, однако их широкое применение сдерживается высокой себестоимостью [35–37].
Поскольку важным компонентом диализного амилоидоза является макрофагальное воспаление, большое значение имеет использование высокоочищенных растворов для приготовления диализата. G. Lonnemann и другие исследователи сообщают о том, что применение ультрачистых диализных жидкостей во время лечения конвективными методами позволяет снизить частоту синдрома запястного канала на 50% [37, 38].
В качестве симптоматической терапии больным диализным амилоидозом может быть рекомендовано местное применение стероидных мазей при болях, в литературе обсуждаются также показания к применению стероидов внутрь и в инъекциях, однако их использование ограничено из-за тяжелой ренальной остеодистрофии. В этой связи представляется перспективным применение у таких больных современных препаратов, подавляющих эффекты фактора некроза опухоли, интерлейкина-1 и интерлейкина-6.
Средством выбора для лечения АTTR-амилоидоза служит трансплантация печени, при которой удается удалить источник синтеза амилоидогенного предшественника. Трансплантация печени существенно задерживает прогрессирование амилоидоза, в первую очередь нервной системы [39]. В то же время накопленные депозиты амилоида в тканях, в особенности в сердце, способны адсорбировать также и нормальный транстиретин и тем самым способствовать дальнейшему прогрессированию амилоидоза [6]. В этой связи в настоящее время широко обсуждаются возможности стабилизации нормального транстиретина и предотвращения его амилоидогенности с помощью фармакологических препаратов (diflunisal) [40].
Поскольку ХПН служит одной из основных причин смерти больных системным амилоидозом, проведение гемодиализа или постоянного амбулаторного перитонеального диализа позволяет улучшить прогноз этих пациентов, в частности, выживаемость этих больных связывают с уровнем бета-2-микроглобулина [41–43].
Выживаемость больных амилоидозом при проведении гемодиализа, независимо от типа амилоидоза, сопоставима с выживаемостью больных другими системными заболеваниями и сахарным диабетом. Постоянный амбулаторный перитонеальный диализ имеет некоторые преимущества перед гемодиализом, поскольку нет необходимости в постоянном сосудистом доступе, отсутствует артериальная гипотония во время процедуры диализа, а у больных с AL-типом амилоидоза во время процедуры возможно удаление легких цепей иммуноглобулинов.
Трансплантация почки одинаково эффективна при обоих типах системного амилоидоза. Пятилетняя выживаемость больных и трансплантата составляет 65 и 62% соответственно и сопоставима с таковыми показателями других групп больных с ХПН. Трансплантация почки показана больным с медленным прогрессированием амилоидоза без поражения сердца и ЖКТ. Амилоидоз в трансплантированной почке возникает, по разным данным, примерно у 30% больных, однако он служит причиной потери трансплантата всего у 2–3% пациентов.
Прогноз при амилоидозе
Амилоидоз характеризуется неуклонно прогрессирующим течением. Прогноз заболевания зависит от типа амилоида, степени вовлечения различных органов, главным образом сердца и почек, наличия и характера предрасполагающего заболевания.
Наиболее серьезен прогноз при AL-амилоидозе. По данным клиники Мейо, средняя продолжительность жизни больных этим типом амилоидоза составляет лишь 13,2 мес., 5-летняя выживаемость – 7%, 10-летняя – всего 1%. При этом самая низкая продолжительность жизни отмечена у пациентов с застойной недостаточностью кровообращения (6 мес.) и ортостатической артериальной гипотонией (8 мес.) [2, 44, 45]. Наиболее частыми причинами смерти больных с AL-типом амилоидоза бывают сердечная недостаточность и нарушения ритма сердца (48%), уремия (15%), сепсис и инфекции (8%). Несмотря на то что смерть от уремии отмечают значительно реже, чем от кардиальных причин, ХПН разной степени выраженности регистрируют более чем у 60% умерших.
Прогноз при АА-амилоидозе более благоприятен и зависит, главным образом, от характера предрасполагающего заболевания и возможности его контроля. Средняя продолжительность жизни больных с этим типом амилоидоза от момента верификации диагноза составляет 30–60 мес. (большая при вторичном амилоидозе, меньшая при амилоидозе в рамках периодической болезни). Эффективное лечение предрасполагающих заболеваний, в том числе полное излечение туберкулеза или хронических нагноений, не исключает возможности развития амилоидоза в дальнейшем, однако замедляет его прогрессирование, улучшая прогноз. Эффективная терапия ревматоидного артрита позволяет продлить течение амилоидной нефропатии, замедляя наступление ХПН. Основной причиной смерти больных АА-типом амилоидоза служит почечная недостаточность.
Достижения последних лет в изучении проблемы амилоидоза, позволившие сформулировать четкие критерии классификации клинических форм амилоидоза и подходы к лечению, дали возможность существенно улучшить прогноз больных разными типами амилоидоза.
Клинические рекомендации по диагностике и лечению системного амилоидоза. Клиническая фармакология и терапия
Л.В. Лысенко (Козловская), В.В. Рамеев, С.В. Моисеев, О.В. Благова, Э.И. Богданов, Г.Е. Гендлин, Д.А. Гришина, А.Я. Гудкова, Е.В. Захарова, О.Е. Зиновьева, О.М. Моисеева, С.С. Никитин, В.А. Парфенов, Н.А. Супонева, С.Н. Терещенко
- DOI
- 10.32756/ 0869-5490-2020-1-13-24
- Количество просмотров
- 9694
В клинических рекомендациях, подготовленных специалистами различного профиля, рассматриваются методы диагностики и лечениясистемного амилоидоза, в том числе АА (вто-ричный амилоидоз при хронических воспалительных заболеваниях, включая ревматоидныйартрит, анкилозирующий спондилит, аутовоспалительные заболевания, хроническиенагноения, злокачественные опухоли и др.), AL (амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема) иATTR (транстиретиновый; семейные формыполиневропатического, кардиопатического идругого амилоидоза, системный старческийамилоидоз). Диагноз амилоидоза, которыйможно заподозрить на основании клиническихданных, необходимо подтвердить при гистологическом исследовании (окрашивание препаратов ткани конго-красным с последующей микроскопией в поляризованном свете). Чтобы замедлить или приостановить прогрессирование амилоидоза любого типа, необходимо добиться уменьшения количества (или, если возможно, удаления) белков-предшественников путем лечения хронического воспаленияпри АА-амилоидозе или подавления пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуно-глобулинов при AL-амилоидозе. Для замедления прогресирования ATTR-амилоидоза упациентов с полиневропатией применяют тафамидис, который ингибирует диссоциацию мутантного транстиретина и снижает его амилоидогенность.
Определение, классификация, группы риска и принципы диагностики
Амилоидоз – группа заболеваний, отличительным признаком которых является отложение в тканях и органах фибриллярного гликопротеида амилоида. Специфическое свойство амилоида, отличающее его от других фибриллярных белков стромы, – способность к двойному лучепреломлению, что проявляется свечением в поляризованном свете предварительно окрашенных конгокрасным препаратов амилоида с изменением красного цвета конгофильных амилоидных отложений на яблочно-зеленый (дихроизм).
В основе амилоидогенеза лежит синтез большого количества нестабильных белковпредшественников, которые агрегируются с образованием амилоидной фибриллы. Клю чевое значение имеет амилоидогенность основного белка-предшественника амилоида, специфичного для каждой формы амилоидоза (в настоящее время известно более 30 таких белков), обозначение которого положено в основу современной классификации заболевания (ВОЗ, 2016 г.). Названия типов амилоида включают в себя букву А, означающую “амилоид», и обозначение конкретного фибриллярного белка амилоида – А (амилоидный А-протеин), L (легкие цепи иммуноглобулинов), TTR (транстиретин), β2М (β2-микроглобулин), В (В-протеин), IAPP (островковый амилоидный полипептид). Используют также производные наименования – иммуноглобулиновый амилоидоз (AL), транстиретиновый (ATTR) и др. (табл. 1) [1-3]. Следует отметить, что Международная классификация болезней (МКБ) 10-го пересмотра базируется на клиническом принципе, не учитывает особенности патогенеза различных форм амилоидоза и не позволяет обосновать адекватное лечение.
| Белок амилоида | Белок-Белок-предшественник | Клиническая форма амилоидоза |
|---|---|---|
| АА | SSA-белок | Вторичный амилоидоз при хронических воспалительных заболеваниях, в том числе периодической болезни и синдроме Макла-Уэллса |
| AL | λ, κ-легкие цепи иммуноглобулинов | Амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема |
| ATTR | Транстиретин | Семейные формы полиневропатического, кардиопатического и др. амилоидоза, системный старческий амилоидоз |
| Аβ2М | β2-микроглобулин | Диализный амилоидоз |
| AGel | Гелсолин | Финская семейная амилоидная полиневропатия |
| AApoAI | Аполипопротеин А-I | Амилоидная полиневропатия (III тип по van Allen, 1956 г.) |
| AFib | Фибриноген | Амилоидная нефропатия |
| Aβ2 | β-белок | Болезнь Альцгеймера, синдром Дауна, наследственные кровоизлияния в мозг с амилоидозом |
| APrPScr | Прионовый белок | Болезнь Крейтцфельда-Якоба, болезнь Герстманна-Штраусслера-Шейнкера |
| AANF | Предсердный натрийуретический фактор | Изолированный амилоидоз предсердий |
| AIAPP | Амилин | Изолированный амилоидоз в островках Лангерганса при сахарном диабете 2 типа, инсулиноме |
| ACal | Прокальцитонин | При медуллярном раке щитовидной железы |
| ACys | Цистатин С | Наследственные кровоизлияния в мозг с амилоидозом (Исландия) |
АА-амилоидоз чаще всего развивается при ревматоидном артрите, серонегативных спондилоартропатиях, аутовоспалительных наследственных периодических лихорадках, в том числе периодической болезни (семейной средиземноморской лихорадке), а также при хронических нагноениях, туберкулезе. АА-амилоид образуется из сывороточного предшественника SAA (serum amyloid A) – острофазового белка, продуцируемого в значительных количествах в ответ на воспаление. По этой причине АА-амилоидоз называют также реактивным или вторичным.
Клинические формы AL-амилоидоза обусловлены единым этиологическим фактором – В-лимфоцитарной дискразией, характеризующейся формированием аномального клона плазматических или В-клеток в костном мозге, которые продуцируют аномальные иммуноглобулины, обладающие амилоидогенностью (легкие цепи моноклонального иммуноглобулина, чаще λ, реже κ-типа). При первичном AL-амилоидозе плазмоклеточная дискразия относительно более доброкачественная, в то время как при В-гемобластозах (множественной миеломе, болезни Вальденстрема и др.) она обладает признаками злокачественной опухоли. Аномальный амилоидогенный клон плазматических клеток может формироваться также из плазмоцитов, локализующихся вне костного мозга, что может привести к развитию локального амилоидоза. Наиболее распространенные локальные формы AL-амилоидоза – амилоидоз трахеи, бронхов и гортани, мочевого пузыря. Выявление плазмоклеточной дискразии необходимо для диагностики AL-амилоидоза, а также для оценки его риска и дифференциального диагноза.
ATTR-амилоидоз является необратимо прогрессирующим заболеванием с высокой степенью инвалидизации вследствие тяжелого поражения сердца, периферической и/или автономной полиневропатии. Пациенты обычно умирают в течение 10-12 лет от первых проявлений. Развитие ATTR-амилоидоза обусловлено мутациями в молекуле транстиретина или возрастным нарушением секреции тетрамеров транстиретина печенью. В обоих случаях происходит распад тетрамеров транстиретина до мономеров, обладающих выраженной конформационной нестабильностью.
Рекомендации:
- Скрининг АА-амилоидоза следует проводить в следующих группах риска: серопозитивные и серонегативные хронические полиартриты (ревматоидный артрит, анкилозирующий спондилоартрит, ювенильный хронический артрит, псориатический артрит, синдром Рейтера и др.), воспалительные заболевания кишечника (болезнь Крона, язвенный колит), аутовоспалительные заболевания (подагра тяжелого рецидивирующего течения, семейные периодические лихорадки – периодическая болезнь, криопиринопатии, TRAPS, гипериммуно глобулинемия D), хронические нагноения (туберкулез, бронхоэктатическая болезнь, остеомиелит и др.), злокачественные солидные опухоли [1,2,4-6].
- Риск АА-амилоидоза у больных с хроническими воспалительными заболеваниями повышается при персистирующем увеличении уровней маркеров острой фазы воспаления (С-реактивный белок, SAA), наличии анемии хронических заболеваний (с повышением уровня ферритина крови), особенно в сочетании с суставным синдромом (синовитом) [4,6-12]. Дина мический контроль за уровнем этих показателей необходим также при мониторировании течения диагностированного АА-амилоидоза. Для оценки риска развития или прогрессирования АА-амилоидоза в условиях преимущественного аутовоспаления или с целью выявления субклинической активации воспаления можно определять сывороточный маркер нейтрофильной активности S100A12 (кальгранулин) [4,13-15].
- Диагностика аутовоспалительных заболеваний предполагает, в первую очередь, проведение генетического исследования на мутации генов MEFV (пирин), NLRP3 (криопирин), TRAPS (рецептор к фактору некроза опухоли альфа), мевалонаткиназы [4,16-25].
- Высокая частота олигосекреторных моноклональных гаммапатий у лиц старше 50 лет требует скринингового обследования этой группы лиц на предмет моноклональных гаммапатий. Наиболее чувствительным и недорогим турбидиметрическим методом для скрининговой диагностики являет Freelite-метод количественной оценки уровня свободных легких цепей иммуноглобулинов. Все больные с плазмоклеточными дискразиями и лимфопролиферативными заболеваниями входят в группу риска AL-амилоидоза [26-34].
- У пациентов с морфологически подтвержденным амилоидозом диагностика AL-амилоидоза предполагает проведение иммунохимического исследования с применением высокочувствительных методов – иммунофиксации сыворотки и суточной мочи, количественного определения свободных легких цепей иммуноглобулинов (Freelite) [26-35].
- Помимо выявления моноклональной гаммапатии, диагностика плазмоклеточной дискразии предполагает выявление и оценку количества плазмоцитов костного мозга, а также их структурных особенностей. Применение цитогенетического исследования и иммунофенотипирования плазмоцитов важно для уточнения клональности и злокачественности аберрантного клона плазматических клеток, в особенности в редких случаях неинформативности иммунохимического исследования крови и суточной мочи [26,33-34,36,37].
- Важное значение в диагностике плазмоклеточных дискразий имеют также иммуногистохимические методы выявления патологического клона плазматических клеток. Это особенно важно для диагностики, типирования и лечения локального варианта ALамилоидоза [35].
- ATTR-амилоидоз следует подозревать у пациентов с полиневропатией, необъяснимым чередованием запоров и диареи, синдромом карпального канала, особенно при наличии полиневропатии, синдрома карпального канала, кардиомиопатии у родственников. Важными симптомами являются геморрагии, фестончатый край зрачка, потеря массы тела, снижение зрения [38-47].
- Диагностика амилоидоза основывается на результатах морфологического исследования [1-2,39,48].
- При системном амилоидозе для диагностики амилоидного поражения органа нет необходимости проводить его биопсию у больных с ранее верифицированным диагнозом амилоидоза по результатам биопсии другого органа. Однако точная диагностика возможна только с помощью морфологического исследования [1,48].
- С целью выявления амилоида необходимо окрашивание препаратов ткани красителем конго-красный с последующей микроскопией в поляризованном свете. Окончательный диагноз амилоидоза устанавливают при выявлении конгофильных масс, обладающих способностью к яблочно-зеленому или желтоватому свечению в поляризованном свете [1,39,48]. Для более точной диагностики амилоидоза применяют также метод окраски тиофлафином Т, который дает светло-зеленое свечение амилоида [1,2].
- При системном амилоидозе информативна биопсия прямой или двенадцатиперстной кишки (с захватом подслизистого слоя). ATTR-амилоид отличается слабой конгофилией. По этой причине выявить этот тип амилоида нередко удается только при повторных биопсиях из разных органов – аспирационной биопсии подкожной жировой клетчатки, биопсии слюнных желез губ и др. Наиболее эффективна биопсия пораженного органа [1-2,39,48]. У пациентов с синдромом запястного канала исследованию на амилоид необходимо подвергать ткань, удаленную при оперативной декомпрессии запястного канала.
- Не рекомендуется проводить биопсию подкожной жировой клетчатки у больных инсулинозависимым сахарным диабетом, так как в местах иньекций инсулин может агрегировать и формировать амилоидные депозиты [48].
- Для дифференциальной диагностики АА-амилоидоза от AL- и ATTR-амилоидоза используют окрасочные методы при тщательном учете клинических предпосылок разных типов амилоидоза [48,49].
- Наиболее эффективным методом типирования является иммуногистохимическое исследование. Поскольку некоторые антисыворотки могут давать перекрестные реакции с разными типами амилоида, исследование целесообразно проводить с панелью антисывороток. Для неспециализированных терапевтических и нефрологических стационаров рекомендуется применение панели антисывороток к SAA, разным типам тяжелых цепей иммуноглобулинов, легким цепям иммуноглобулинов λ и κ, транстиретину. Важно также использовать антисыворотки к фибриногену [36,50].
- Для диагностики ATTR-амилоидоза необходимо генетическое исследование на наличие мутации гена транстиретина [39].
Клинические проявления
Для вторичного АА-амилоидоза характерно более раннее начало, чем для AL-амилоидоза (средний возраст больных составляет около 40 и 65 лет, соответственно). ATTR-амилоидоз, несмотря на наследственную природу, характеризуется низкой пенетрантностью и также проявляется обычно после 35 лет.
Поражение почек – ведущий клинический признак АА- и AL-амилоидоза, наблюдающийся практически у всех больных. Поражение почек встречается и у больных с многими формами семейного амилоидоза (AFib, ALys, AGel и др.). При ATTR-амилоидозе нефропатия отмечается лишь у 20-23% больных. Клинически амилоидная нефропатия характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурия, нефротический синдром, хроническая почечная недостаточность (ХПН). Иногда возможно развитие ХПН без предшествующего нефротического синдрома.
Поражение сердца развивается у подавляющего большинства больных AL-амилоидозом и у 50-60% пациентов с АTTR-амилоидозом, но не характерно для АА-амилоидоза (рис. 1). При эхокардиографии у больных амилоидозом сердца наблюдается утолщение межжелудочковой перегородки и стенки левого желудочка (чаще симметричное), которое не сопровождается электрокардиографическими признаками гипертрофии миокарда. У части больных отмечается снижение вольтажа зубцов на ЭКГ, хотя отсутствие этого признака не исключает диагноз амилоидоза сердца. Нарушение диастолической функции левого желудочка (рестриктивный тип) приводит к развитию сердечной недостаточности, которая быстро прогрессирует, плохо поддается лечению и почти у 50% пациентов оказывается причиной смерти. Кроме того, у больных амилоидозом сердца часто наблюдаются различные аритмии и нарушения проводимости.
Рис. 1. Алгоритм дифференциальной диагностики амилоидоза сердца. *Если предполагается амилоидоз сердца, то скрининговую биопсию (подкожной жировой клетчатки, слизистой оболочки прямой кишки) целесообразно выполнить параллельно с другими исследованиями. При отрицательном результате показана биопсия пораженного органа, включая сердце.При AL-амилоидозе и особенно ATTR-амилоидозе часто встречается ортостатическая артериальная гипотензия – вариант сосудистой недостаточности, при которой сосуды теряют способность поддерживать нормальное артериальное давление в условиях ортостатических нагрузок. Она проявляется ощущением дурноты и потемнением в глазах в ортостазе в сочетании с резким снижением АД. Обычно этот симптом связан с дисфункцией автономной нервной системы (амилоидоз нервных сплетений сосудов). Тяжелая ортостатическая гипотензия сопровождается обмороками, а иногда приводит к развитию острого нарушения мозгового кровообращения.
Поражение желудочно-кишечного тракта может проявляться, особенно при AL-амилоидозе, тяжелой диареей или динамической непроходимостью, которые чаще связаны с нарушениями моторики кишечника вследствие дисфункции автономных нервных сплетений. Иногда выявляют изъязвления или перфорацию стенок с возможным кровотечением. При поражении пищевода возможна дисфагия.
Поражение печени при АА- и AL-типах амилоидоза наблюдают практически в 100% случаев. Функция печени чаще остается сохранной, редким признаком амилоидоза печени является внутрипеченочная портальная гипертензия. При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения.
Увеличение селезенки, обусловленное амилоидным поражением, отмечается у большинства больных и обычно сопутствует увеличению печени.
Поражение нервной системы, представленное симптомами периферической соматической и автономной невропатии, отмечают у 17-35% больных AL-амилоидозом и практически у всех пациентов с наследственной амилоидной полиневропатией разных типов (ATTR, AApoA1 и др.). В большинстве случаев развивается дистальная симметричная полиневропатия с неуклонно прогрессирующим течением, различные дисфункции автономной нервной системы. Реже выявляют двусторонний синдром запястного канала, обусловленный сдавлением срединного нерва депозитами амилоида.
Поражение кожи наблюдают почти у 40% больных AL-амилоидозом. Помимо параорбитальных геморрагий описаны также папулы, бляшки, узелки, пузырьковые высыпания, склеродермоподобная индурация кожи.
Амилоидные отложения в мышцах чаще встречаются при AL-амилоидозе. Макроглоссия – патогномоничный симптом AL-амилоидоза, развивающийся примерно у 20% пациентов.
Редким проявлением амилоидоза, описанным при AL- и, в особенности, АTTR-типах, бывает поражение глаз (сухой кератоконъюнктивит, вторичная глаукома, помутнение стекловидного тела, дисфункции зрачка).
Клиническая картина других типов амилоидоза варьируется в зависимости от основной локализации и распространенности амилоидных депозитов, которые иногда могут быть значительными и напоминать проявления AL-амилоидоза.
Рекомендации:
- Наиболее типичное проявление амилоидоза почек – изолированная протеинурия более 0,5 г/сут, чаще нефротического уровня. Иногда при множественной миеломе важное значение приобретает иммунохимическое электрофоретическое исследование мочи для отличия альбуминурии в рамках амилоидоза и протеинурии переполнения (наличие в моче белка БенсДжонса, реакция термопреципитации белка Бенс-Джонса не обладает достаточной информативностью). Для установления связи протеинурии с амилоидозом необходимо также исключить протеинурию, связанную с диабетической нефропатией и гипертонической почкой [4,5,29,48,51-56].
- На амилоидоз сердца указывает утолщение межжелудочковой перегородки и/или задней стенки левого желудочка более 12 мм при эхокардиографии, особенно в сочетании с низкоамплитудной ЭКГ. Дифференциальный диагноз проводят с гипертрофией левого желудочка, которая может быть следствием артериальной гипертонии, аортальных пороков, гипертрофической кардиомиопатии и других причин [29,37-39,42,48,51-53,57-62].
- Характерное проявление амилоидоза сердца – низкая амплитуда желудочковых комплексов на ЭКГ (менее 5 мм в отведениях от конечностей). Пато логические Q-зубцы у больных амилоидозом нередко являются псевдоинфарктными (вследствие электрически нейтральных отложений амилоида, имитирующих рубцовые изменения), однако при амилоидозе коронарных артерий возможно развитие и истинного инфаркта миокарда [29,37-39,42,48,51-53,56-62].
- При амилоидозе сердца наблюдается рестриктивное нарушение диастолической функции левого желудочка, фракция выброса часто остается нормальной, или степень ее снижения не соответствует тяжести сердечной недостаточности [29,37-39,42,48,51-53,5662].
- Всем больным с амилоидозом сердца необходимо проведение стандартной эхокардиографии с допплерометрической оценкой трансмитрального крово тока, при наличии технической возможности оправдано также проведение тканевой допплерометрии миокарда для более точной оценки внутрисердечной гемодинамики. В стандарт обследования больных амилоидозом сердца входят также ЭКГ и суточное мониторирование АД и ЭКГ [1,2,29,37-39,42,48,5153,56-62].
- Магнитно-резонансная томография (МРТ) с контрастированием гадолинием с высокой вероятностью выявляет инфильтративный характер поражения миокарда и имеет особое диагностическое значение при “изолированном» поражении сердца [48,56,6062].
- Амилоидную инфильтрацию сердца позволяет выявить также сцинтиграфия миокарда с пирофосфатом технеция, особенно при наличии трудностей в морфологической диагностике ATTR-амилоидоза, для которого характерна слабая конгофилия пораженных тканей. Интенсивное накопление радиоактивного препарата в миокарде (2+/3+) в сочетании с утолщением миокарда неясной этиологии указывает на высоко вероятный ATTR-амилоидоз, если у пациента исключен диагноз AL-амилоидоза (рис. 1) [39,42,58-59,62].
- Диагноз амилоидоза сердца может быть подтвержден при биопсии миокарда. Однако проведение этого исследования обычно не требуется при наличии типичных эхокардиографических изменений у пациентов с амилоидозом, установленным при биопсии другого органа, например, почки или слизистой оболочки прямой или двенадцатиперстной кишки.
- Диагностика периферической амилоидной полиневропатии основывается на клинической оценке неврологических проявлений: обычно выявляют различные нарушения чувствительности, в частности температурной и болевой. Из-за поражения преимущественно мелких немиелинизированных волокон электромиография и исследование скорости проведения нервного импульса обычно неинформативны для ранней диагностики амилоидной полиневропатии [38,39]. Доминирование жалоб, связанных с поражением нервной системы, является отличительной чертой ATTR-амилоидоза.
- Электромиография наряду с другими нейрофизиологическими методами (количественное сенсорное тестирование, конфокальная микроскопия нервов роговицы, оценка состояния интраэпидермальных нервных волокон в биоптате кожи) может использоваться для оценки характера (признаки аксонального и/или аксонально-демиелинизирующего типа поражения двигательных и чувствительных волокон нервов конечностей) и тяжести неврологических нарушений. Электромиография в сочетании с ультразвуковым исследованием периферических нервов позволяет объективизировать их повреждение в анатомически узких каналах (туннельные невропатии) [38,39].
- Признаками I стадии амилоидной полиневропатии являются незначительные нарушения чувствительности по полиневропатическому типу, более выраженные в ногах, пациент сохраняет способность к самостоятельной ходьбе. При II стадии полиневропатии из-за нарастания чувствительных и присоединения двигательных нарушений в виде нижнего периферического преимущественно дистального парапареза походка пациента нарушается, требуется опора на трость или костыли. На III стадии тяжелые двигательные нарушения приковывают пациента к постели, передвижение возможно только на инвалидной коляске [38,39,46].
- Поражение вегетативной нервной системы чаще всего проявляется ортостатической гипотензией разной степени тяжести. Однако систолическое артериальное давление менее 90 мм рт. ст. может быть обусловлено низким сердечным выбросом у больных с сердечной недостаточностью или гиповолемией при тяжелом нефротическом синдроме. Другие частые проявления поражения вегетативной нервной системы – моторная диарея, дисфункция мочевого пузыря, половой сферы, гипогидроз [1,2,38,51]. Наряду с моторной диареей причиной значительного (на 9-18 кг) снижения массы тела могут быть нарушения трофики мышц [38,39].
- Двусторонний синдром запястного канала наиболее характерен для ATTR-, β2М- и AL-амилоидоза и проявляется интенсивными болями и парестезиями в IIII пальцах кистей с постепенной атрофией мышц тенара [38,39].
- МРТ головного мозга с контрастным усилением проводят при наличии клинических признаков поражения центральной нервной системы [38,39].
- Основным признаком амилоидоза печени является ее увеличение, наиболее специфична гепатомегалия более 15 см по данным компьютерной томографии. У больных амилоидозом печени обычно выявляют также холестаз (повышение активности щелочной фосфатазы и/или g-глютамилтранспептидазы в 1,5 раза по сравнению с верхней границей нормы). Ложная диагностика амилоидоза печени возможна у больных с тяжелой застойной правожелудочковой недостаточностью [2,4,48,51].
- Для выявления амилоидоза печени, селезенки и почек всем пациентам проводят ультразвуковое исследование этих органов, в некоторых случаях необходимо проведение компьютерной томографии брюшной полости [2,48].
- Диарея вследствие инфильтрации амилоидом стенки желудочно-кишечного тракта возникает редко, такую диарею трудно дифференцировать от моторной диареи в рамках поражения вегетативной нервной системы. Наиболее надежно вовлечение желудочнокишечного тракта при амилоидозе устанавливают по результатам морфологического исследования. Одна ко обнаружение амилоида только в стенках сосудов желудочно-кишечного тракте не является критерием его поражения, необходимо обнаружение амилоидных депозитов в интерстиции подслизистого слоя кишечника [1-2,5,48,52].
- Нодулярный легочный и трахеобронхиальный амилоидоз за редким исключением – это проявление локального AL-амилоидоза. Для системного AL-амилоидоза характерно обнаружение диффузного интерстициального легочного амилоидоза. В связи с редкостью дыхательной недостаточности необходимости в морфологической верификации легочного амилоидоза обычно не возникает. Наиболее информативным методом диагностики амилоидоза легких является компьютерная томография [1,2,48]. У больных локальным трахеобронхиальным AL-амилоидозом важными методами мониторирования течения заболевания являются ларингоскопия и бронхоскопия [27,48].
- На амилоидоз плевры указывает рецидивирующий плевральный выпот, который не зависит от эффективности лечения отечного синдрома, обусловленного сердечной недостаточностью или нефротическим синдромом. При амилоидном поражении плевры жидкость, полученная во время пункции плевральной полости, нередко содержит примесь крови. При амилоидозе плевры эвакуация плеврального выпота, как правило, малоэффективна из-за быстрого его накопления [1,2,48].
- Поражение мягких тканей характерно для AL-амилоидоза. Макроглоссия с инфильтрацией дна ротовой полости и периорбитальная пурпура (и кожные геморрагии на теле) патогномоничны для этого типа амилоидоза. Возможны также псевдогипертрофия скелетных мышц с развитием мышечной слабости, лимфаденопатия, амилоидоз височной артерии [1,2,39,48,52].
- Признаками прогрессирования амилоидоза сердца являются дальнейшее утолщение миокарда (на 2 мм и более), увеличение функционального класса сердечной недостаточности, снижение фракции выброса левого желудочка на 10% и более. Показатели тяжести амилоидоза сердца – увеличение уровня NTproBNP (особенно более 1800 нг/л) и тропонинов (тропонин Т более 0,025 нг/мл). Критериями прогрессирования амилоидоза почек считают увеличение протеинурии (на 50% от исходного уровня, как правило на 1 г/сут и более) и сывороточного уровня креатинина (на 25% и более от исходного). Информативным критерием прогрессирования амилоидоза печени является увеличение активности щелочной фосфатазы на 50% от исходной. Прогрес сирование полиневропатии объективизируют на основании результатов стимуляционной электромиографии и других нейрофизиологических и нейровизуализационных методов обследования (см. выше). Важным показателем тяжести больных AL-амилоидозом является разница в содержании свободных легких цепей иммуноглобулинов более 180 мг/л, установленная методом Freelite [2,38,51,48,60,61,64].
Лечение системного амилоидоза
Целью терапии любого типа амилоидоза служит уменьшение количества (или, если возможно, удаление) белков-предшественников для того, чтобы замедлить или приостановить прогрессирование болезни. Неблаго приятный прогноз при естественном течении амилоидоза оправдывает применение агрессивных методов лечения. Клиническое улучшение, достигаемое с помощью лечения, включает стабилизацию или восстановление функции жизненно важных органов, а также предотвращение функциональных нарушений с увеличением продолжительности жизни больных. Лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности сердечной недостаточности, аритмии, отечного синдрома, коррекцию артериальной гипотензии и др.
Лечение АА-амилоидоза
Цель терапии АА-амилоидоза – подавление продукции белка-предшественника SAA (вплоть до устойчивой нормализации), что достигается активным лечением хронического воспаления (в том числе субклинического). Это позволяет уменьшить клинические проявления и предотвратить прогрессирование амилоидной нефропатии и существенно улучшить прогноз.
Рекомендации:
- Основной стратегией лечения АА-амилоидоза является эффективное подавление воспаления. Лечение должно проводиться вне зависимости от клинической активности воспалительного заболевания до нормализации уровня маркеров острой фазы воспаления – С-реактивного белка (предпочтительно применение высокочувствительного метода измерения) и/или SAA [10-13,63,66-77].
- Больным ревматоидным артритом и серонегативными спондилоартропатиями необходима постоянная пожизненная базисная терапия. Предпочтение отдают генно-инженерным биологическим препаратам, в том числе ингибиторам фактора некроза опухоли-α, интерлейкина-6, ритуксимабу. После оценки эффективности и безопасности базисной терапии оправдано присоединение терапии колхицином в дозе 2 мг/сут [10-13,63,66-77].
- Препаратом выбора при периодической болезни и тяжелой рецидивирующей подагре является колхицин в дозе 2 мг/сут. Начальная доза препарата составляет 0,5 мг/сут, затем ее постепенно увеличивают до целевой под контролем клинического анализа крови и сывороточного уровня креатинина. Для предупреждения осмотической диареи, обусловленной колхицином, возможно временное назначение ферментных препаратов. Дозу колхицина снижают до 1 мг/сут у больных с хронической болезнью почек 4-5 стадии [1,2,4,69]. При неэффективности колхицина показано назначение ингибиторов интерлейкина-1, в частности канакинумаба.
- При криопиринопатиях, TRAPS препаратами выбора являются ингибиторы интерлейкина-1, к которым присоединяют колхицин [18,19,74-77].
- При хронических нагноениях важное значение имеет хирургическое лечение. В дальнейшем или одновременно проводят лечение димексидом (5-10 г/сут в разведении большим количеством соков – томатного, гранатового и др.). Препарат предпочтителен при легочных нагноениях [1-2,4,6,31].
Лечение АL-амилоидоза
При AL-амилоидозе, как и при множественной миеломе, целью лечения служит подавление пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуноглобулинов. В отличие от множественной миеломы, принципиальной задачей лечения AL-амилоидоза является по возможности полная элиминация патологического клона. В связи с быстрым прогрессированием заболевания важное значение имеет применение быстродействующих схем лечения на основе бортезомиба. По мере достижения ремиссии у некоторых больных применяют высокодозную химиотерапию с поддержкой аутологичными стволовыми клетками. При строгом подборе больных с исключением противопоказаний к этой терапии эффект достигают у 60% больных. У больных с клиническими симптомами амилоидоза сердца, ортостатической гипотензией, диареей, желудочно-кишечными кровотечениями в анамнезе, а также у лиц старше 70 лет с амилоидным поражением двух и более систем организма проведение высокодозной химиотерапии не рекомендуется. Тяжелый агранулоцитоз и другие осложнения существенно ограничивают ее применение. Проводят также лечение талидомидом или леналидомидом. Колхицин при AL-амилоидозе не эффективен.
Рекомендации:
- Основной стратегией лечения АL-амилоидоза является элиминация амилоидогенного клона плазматических клеток костного мозга. После достижения гематологической ремиссии проводят противорецидивное лечение в течение не менее 12 месяцев. В этот период при отсутствии противопоказаний возможна высокодозная химиотерапия с поддержкой аутологичными стволовыми клетками, которая позволяет достичь длительной ремиссии [2,26,37,56,57,65,78,79].
- Полный гематологический ответ диагностируют на основании исчезновения моноклональных амилоидогенных иммуноглобулинов по данным иммунофиксации крови и суточной мочи, количественного определения свободных легких цепей иммуноглобулинов методом Freelite (нормальный уровень легких цепей κ – менее 19,4 мг/л, λ – менее 26,3 мг/л, нормальное соотношение легких цепей в пределах 0,261,65). При снижении уровня свободных легких цепей на 50% от исходного диагностируют частичный ответ, критерием очень хорошего частичного ответа является разница между содержанием двух типов легких цепей иммуноглобулинов менее 40 мг/л [37,60,61,64].
- Клинический эффект терапии в первую очередь оценивают по динамике кардиологических и ренальных показателей. Особо выделяют NT-proBNP-ответ (снижение уровня маркера на 30% и более или на 300 нг/л и более у пациентов с исходным уровнем более 650 нг/л). С клиническим эффектом лечения больше коррелирует почечный ответ (снижение протеинурии на 75% и более, повышение сывороточного креатинина не более 25% от исходного). Ответ со стороны других органов не обладает существенной прогностической информативностью. Эффективность лечения амилоидоза печени оценивают по снижению активности щелочной фосфатазы (на 50% и более) и уменьшению размеров печени (краниокаудальный размер по данным КТ должен уменьшиться на 30% в течение года после достижения гематологической ремиссии). Эффективность лечения амилоидной полиневропатии определяют, главным образом, по результатам клинического неврологического осмот ра. Уменьшение амилоидных депозитов в мягких тканях может быть оценено по данным компьютерной томографии или МРТ. Эффективным методом оценки общего содержания амилоида в тканях служит сцинтиграфия с радиоактивным амилоидным Ркомпонентом [2,26,37,57,60,61,64].
- Учитывая возможность достижения быстрого гематологического ответа, терапией первой линии, особенно у больных с высоким риском быстрого прогрессирования, считают комбинированные схемы на основе бортезомиба, например, трехкомпонентная схема: бортезомиб 1,3 мг/м2 внутривенно или подкожно (1, 5, 8 и 11-й дни цикла), мелфалан 0,15 мг/кг внутрь (с 1-го по 4-й день) и дексаметазон 20 мг/сут внутрь (1, 5, 8 и 11-й дни). Для большей безопасности предпочтительно подкожное введение бортезомиба. Если в дальнейшем планируется высокодозная химиотерапия с поддержкой аутологичными стволовыми клетками мелфалан в составе трехкомпонентной схемы заменяют на циклофосфамид (400 мг внутривенно капельно в 1, 8, 12-й дни), который не вызывает истощение пула стволовых клеток в костном мозге. Курсы химиотерапии на основе бортезомиба проводят каждые 4 недели (всего 8 курсов). Одновременно назначают омепразол, низкомолекулярные гепарины (для профилактики тромбозов при применении высоких доз дексаметазона), при наличии показаний – антибиотики, противогрибковые препараты, ацикловир [65,78,79].
- Не менее эффективна схема терапии мелфаланом (внутрь 0,15 мг/кг с 1-го по 4-й день) и дексаметазоном (внутрь 20 мг/сут в дни 1-4, 9-12 и 17-21) каждые 4-6 недель. Основным недостатком этой схемы является медленное формирование гематологического ответа, что делает ее менее перспективной у больных с высоким риском быстрого прогрессирования амилоидоза. Одновременно назначают омепразол, низкомолекулярные гепарины (для профилактики тромбозов при применении высоких доз дексаметазона), при наличии показаний – антибиотики, противогрибковые препараты, ацикловир [1,2,37,56,57,65,78,79].
- У тяжелых больных с декомпенсированной сердечной недостаточностью может применяться схема терапии мелфаланом (внутрь 0,15 мг/кг с 1-го по 4-й дни) и преднизолоном (0,8 мг/кг с 1-го по 7-й день). Однако эффективность этой схемы ограничена [1,2,56,57,65,78,79].
Лечение ATTR-амилоидоза
До недавнего времени единственным методом лечения ATTR-амилоидоза была трансплантация печени, секретирующей нормальный транстиретин. Поскольку 98% всего сывороточного транстиретина синтезируется печенью, это позволяло прервать продукцию мутантного транстиретина. Трансплантация печени существенно замедляет прогрессирование ATTR-амилоидоза, а 20летняя выживаемость больных после трансплантации составляла 55,3% [36]. Однако уже имеющиеся массы амилоида способны выступать в роли ядра нуклеации для новых депозитов амилоида на основе нормального транстиретина (амилоидускоряющая субстанция). В настоящее время у больных с ранними стадиями ATTRамилоидной полиневропатии апробированы консервативные методы стабилизации тетрамерной структуры мутантного транстиретина и, следовательно, подавления его амилоидогенности. Один из таких препаратов – тафамидис замедлял на 52% (р=0,027) прогрессиро вание неврологических нарушений у больных ATTRамилоидозом, сохраняя функцию периферических соматических и автономных нервных волокон [12]. В клиническом исследовании III фазы лечение тафамидисом по сравнению с плацебо у больных с ATTR-амилоидозом сердца вызывало снижение общей смертности и частоты госпитализаций по сердечно-сосудистым причинам и задерживало ухудшение функциональной активности [81]. В Российской Федерации применение тафамидиса зарегистрировано только для лечения ATTR-амилоидоза с периферической полиневропатией.
В качестве стабилизатора транстиретина изучается также дифлюнизал из группы нестероидных противовоспалительных препаратов, однако его эффективность показана только в экспериментальных условиях.
Рекомендации:
- Трансплантация печени остается одним из методов лечения ATTR-амилоидоза. Наилучших показателей выживаемости удается достичь у больных моложе 50 лет с небольшой длительностью заболевания, нормальным индексом массы тела и мутацией Val30Met. Эффективность трансплантации печени существенно ниже у больных с III стадией полиневропатии, ортостатической гипотензией, хронической сердечной недостаточностью, удлинением комплекса QRS бо лее 120 мс и утолщением межжелудочковой перегородки [2,38-40,43-46].
- Больным ATTR-амилоидозом с периферической невропатией рекомендуется пожизненный прием тафамидиса в дозе 20 мг/сут внутрь, который обладает хорошим профилем безопасности [38-40,43-46,80].
Заместительная почечная терапия
Рекомендации:
- Поскольку ХПН служит одной из основных причин смерти больных системным амилоидозом, проведение гемодиализа или постоянного амбулаторного перитонеального диализа позволяет улучшить прогноз этих пациентов. Выживаемость больных амилоидозом при проведении гемодиализа, независимо от его типа, сопоставима с выживаемостью больных другими системными заболеваниями и сахарным диабетом. При этом хорошую и удовлетворительную реабилитацию отмечают у 60% пациентов с АА- и AL-типами амилоидоза. Основной причиной смерти больных амилоидозом при проведении гемодиализа являются сердечно-сосудистые осложнения. Амбула торный перитонеальный диализ имеет некоторые преимущества перед гемодиализом, учитывая отсутствие необходимости в постоянном сосудистом доступе и меньший риск артериальной гипотензии во время процедуры диализа. У больных AL-амилоидозом во время процедуры возможно удаление легких цепей иммуноглобулинов [1-4,6,28,48].
- Трансплантация почки эффективна при системном амилоидозе: 5-летняя выживаемость больных и трансплантата составляет 65% и 62%, соответственно, и сопоставима с таковыми в других группах больных с ХПН. Трансплантация почки показана больным с медленным прогрессированием амилоидоза без поражения сердца и желудочно-кишечного тракта. Амилоидоз в трансплантированной почке возникает, по разным данным, примерно у 30% больных, однако он служит причиной потери трансплантата всего у 2-3% пациентов [1-4,6,28,48].
Симптоматическая терапия
Рекомендации:
- Больным амилоидозом противопоказаны сердечные гликозиды и недигидропиридиновые антагонисты кальция (верапамил, дилтиазем), которые могут накапливаться в амилоиде, в то время как β-адреноблокаторы и ингибиторы АПФ следует применять осторожно. Основа лечения застойной сердечной недостаточности при амилоидозе сердца – петлевые диуретики, хотя ортостатическая гипотензия может ограничивать применение этих препаратов, как и β-адреноблокаторов и ингибиторов АПФ [1,2,31,45,49,52,58].
- При наличии желудочковых тахиаритмий, сопровождавющихся высоким риском внезапной сердечной смерти, показана установка искусственного кардиовертера-дефибриллятора, а при синдроме слабости синусового узла и атрио-вентрикулярной блокаде – имплантация искусственного водителя ритма [2,38,39,45,52,53,58].
- Для лечения ортостатической гипотензии могут быть использованы минералокортикостероиды или глюкокортикостероиды, хотя их применение может привести к декомпенсации сердечной недостаточности. Возможно назначение α-адреномиметика мидодрина (Гутрон), однако его применение требует осторожности [1,2,39,42,45,52,58].
- Для контроля кишечной моторики высокую эффективность показали пролонгированные препараты соматостатина (октреотид-лонг 20 мг один раз в месяц) [1,2,38,39,49].
- При деструкции стекловидного тела у больных ATTR-амилоидозом может потребоваться витрэктомия. При наличии неконтролируемой глаукомы проводят трабекулэктомию [39].
- Для купирования невропатической боли у пациентов с амилоидной невропатией рекомендовано использование адъювантных анальгетиков (антиконвульсанты и антидепрессанты) [39].
причины, симптомы, лечение и профилактика
Амилоидоз почек — это заболевание, при котором в тканях почек происходит нарушение белково-углеводного обмена и откладывается особый нерастворимый белок — амилоид. Это приводит к развитию некротического синдрома, нарушению функции почек и хронической почечной недостаточности.
На долю амилоидоза почек приходится около 3% всех почечных заболеваний.
Классификация форм амилоидоза почек
В зависимости от механизма заболевания и причин амилоидоза в современной урологии выделяют следующие формы амилоидоза почек: приобретенную, идиопатическую, семейную, старческую, а также локальную опухолевую.
При первичном, или идиопатическом амилоидозе почек, причина болезни и ее механизм остаются невыясненными. Вторичная или приобретенная форма амилоидоза почек развивается вследствие различных иммунологических нарушений (ревматических болезней, хронических инфекций, злокачественных опухолей). Вторичная форма амилоидоза является наиболее распространенной.
Семейный или наследственный амилоидоз почки передается по наследству. Он обусловлен генетическим дефектом образования белков в организме.
Старческий амилоидоз почек возникает в пожилом возрасте. Природа развития локального амилоидоза почек пока не ясна.
Первичная, семейная локальная и старческая формы амилоидоза почек являются самостоятельными заболеваниями, а вторичный амилоидоз считается осложнением основного заболевания.
В зависимости от того, какой тип фибриллярного белка содержится в амилоиде, различают следующие типы амилоидоза:
- АА-тип — вторичный амилоидоз, при котором в амилоиде содержится сывороточный альфа-глобулин.
- AL-тип — первичный амилоидоз, в амилоиде — легкие цепочки lg.
- ATTR-тип (семейный или старческий амилоидоз, в амилоиде — белок транстиретин).
В зависимости от того, поражение какого органа преобладает, различаются амилоидоз нефропатический, гепатопатический, энтеропатический, кардиопатический, нейропатический, панкреатический и смешанный. Независимо от формы амилоидоза и его типа в тканях происходит вытеснение амилоидным веществом элементов тканей, понижение и последующая утрата функций отдельных органов.
Прогноз и профилактика амилоидоза почек
Прогноз амилоидоза почек во многом зависит от скорости прогрессирования амилоидоза почек и течения основного заболевания. Если отложения амилоида носят локальный характер, а клинические проявления болезни выражаются слабо, то амилоидоз не представляет угрозы для жизни пациента.
Прогноз ухудшается, если к амилоидозу почек присоединяются инфекции, кровоизлияния, развитие тромбозов. Сказывается на развитии амилоидоза почек и пожилой возраст. Если у пациента развивается сердечная и почечная недостаточность, то выживаемость составляет меньше года. Успешному выздоровлению способствует своевременное обращение к врачу, ранняя диагностика амилоидоза, активное лечение и окончательное устранение основного заболевания.
Для профилактики амилоидоза почек необходимо своевременное лечение любой хронической патологии, которая может привести к развитию заболевания. В качестве его профилактики необходимо отказаться от употребления алкоголя и курения, правильно питаться и вести здоровый образ жизни. Кроме того, необходимо избегать контакта с токсическими веществами, а также необоснованного приема лекарственных препаратов.
Причины развития амилоидоза почек
Каждая форма амилоидоза почек имеет свои причины. Этиология первичного амилоидоза почек чаще всего остается неизвестной, иногда болезнь может развиваться при множественной миеломе. При идиопатическом амилоидозе кроме почек могут поражаться кожа, язык, кишечник, печень, легкие и сердце.
Вторичный амилоидоз почек связан с системными заболеваниями, опухолями, хроническими инфекциями. При вторичной форме амилоидоза поражаются почки, печень, лимфоузлы, сосуды.
Старческий амилоидоз почек является признаком старения и встречается у 80% людей в возрасте старше 80-ти лет. Семейный амилоидоз почек развивается при периодической болезни, чаще всего встречается у жителей стран средиземноморского бассейна. Причиной локальной формы амилоидоза почек могут стать болезнь Альцгеймера, опухоли эндокринной системы, сахарный диабет. При локальном амилоидозе узелковые образования могут появляться в гортани, легких, на коже, языке, в мочевом пузыре. Диализный амилоидоз в настоящее время является осложнением хронической почечной недостаточности. Развивается такое осложнение у больного, длительное время находящегося на гемодиализе.
На сегодняшний день существуют и другие гипотезы возникновения амилоидоза почек: иммунологическая, мутационная.
Симптомы амилоидоза почек
Выделяют четыре стадии в развитии амилоидоза почек, и на каждой из них амилоидоз почек имеет разные симптомы. Латентная стадия амилоидоза почек, несмотря на наличие амилоида в почках, протекает практически бессимптомно. Для нарушения функций количество амилоида в почках еще недостаточно. В этот период могут возникать симптомы, характерные для первичного заболевания (ревматических болезней, инфекций). УЗИ не выявляет никаких изменений в почках, в крови может быть немного повышен холестерин, а анализ мочи периодически показывает повышенный белок. Латентная стадия амилоидоза почек может длиться до 3-5-ти лет и даже больше. Если сделать пункцию, то на этой стадии болезни уже можно выявить амилоид в трубочках, клубочках и почечных канальцах. Но болезнь никак себя не проявляет, а делать биопсию без причины никто не станет, поэтому амилоидоз почек плавно переходит в протеинурическую стадию.
В данном состоянии основным проявлением амилоидоза почек является постоянная протеинурия. Для нее характерны колебания белка в моче от 0,1 до 3,0 г/л. Как правило, выявляется это случайно, при анализах на первичной стадии заболевания. Кроме того, на этой стадии амилоидоза почек наблюдается проявление микрогематурии, лейкоцитурии, повышение СОЭ. Амилоид в почках продолжает накапливаться, набирают силу дегенеративные изменения, меняется биохимический состав крови, появляется анемия. Процесс может длиться 10-15 лет. Почки увеличиваются в размерах, становятся плотными, приобретают серо-розовый матовый оттенок.
При нефротической стадии амилоидоза почек на первый план выходит нефротический синдром. Уровень белка в моче резко повышается, а в крови — снижается. Больной худеет, постоянно жалуется на повышенную жажду, тошноту, слабость. Артериальное давление постоянно меняется, может возникать предобморочное состояние. Амилоид накапливается в кишечнике и приводит к диарее. Любое заболевание, например, грипп, ангина и др., приводят к развитию отеков в конечностях, на лице. Отеки могут распространяться и на другие органы: легкие, брюшную полость, сердце. Отложение амилоида в печени, почках, селезенке вызывает их уплотнение.
На уремической стадии амилоидоза почек возникает тяжелая форма почечной недостаточности, истощение организма. Почка сморщивается, уплотняется и перестает выполнять свои функции, выделение мочи полностью прекращается. На этой стадии стойкие отеки при амилоидозе почек сохраняются. Кроме того, амилоидоз может осложняться тромбозом почечных вен с болевым синдромом. Все это может привести к летальному исходу.
При амилоидозе могут наблюдаться не только почечные, но и внепочечные симптомы. Системными проявлениями амилоидоза являются слабость, одышка, аритмия, головокружение, анемия. Если к заболеванию присоединяется амилоидоз кишечника, то у пациента развивается стойкая диарея.
Обнаружили симптомы данного заболевания?Звоните
Наши специалисты проконсультируют Вас!
Диагностика амилоидоза почек
Диагностика амилоидоза представляет собой непростую задачу. На ранних стадиях причины амилоидоза почек не ясны, да и само проявление заболевания может быть нечетким, а при вторичном амилоидозе симптомы основного хронического заболевания «смазывают» всю клиническую картину.
Если у пациента наблюдаются такие симптомы, как сердечная, почечная недостаточность, поражение нервной системы, белок в моче, увеличение селезенки и печени, то врач может предположить развитие амилоидоза. В данной ситуации не обойтись без лабораторных исследований крови и мочи. Общий анализ мочи позволяет выявить нарастающую протеинурию, микрогематурию, лейкоцитурию. Общий анализ крови поможет выявить анемию, лейкоцитоз, повышение СОЭ. Показательным может быть и проведение копрограммы. Она позволяет выявить значительное содержание мышечных волокон в испражнениях, большое количество крахмала в кале.
Достоверным методом при диагностике амилоидоза почек является биопсия почки. При морфологическом исследовании биоптата после окраски его конго красным в поляризованном свете можно выявить зеленое свечение, характерное для амилоидоза почек. Амилоид может быть выявлен в клубочках, по ходу канальцев, сосудов. В ряде случаев при амилоидозе почек проводят биопсию кожи, десны, печени, слизистой прямой кишки.
Для диагностики амилоидоза почек применяют и ультразвуковое исследование, позволяющее выявить увеличение почек, печени, селезенки. Довольно часто для диагностики амилоидоза почек используют ЭКГ и рентгенографию ЖКТ.
Лечение амилоидоза почек
Во время лечения вторичного амилоидоза почек очень важна успешность терапии основного заболевания. Если первичная патология будет излечена, то и симптомы амилоидоза почек могут регрессировать.
Патогенетическое лечение амилоидоза почки заключается в подавлении образования амилоида. Для этого необходимо в течение 6-12 месяцев ежедневно употреблять в пищу 100-150 г сырой печени, белки и антиоксиданты, которые значительно тормозят развитие амилоидоза почек. Для этой цели также применяют печеночные гидролизаты. Наряду с этим при амилоидозе почек назначаются препараты, тормозящие синтез амилоида. Симптоматическое лечение при амилоидозе почек включает в себя назначение гипотензивных препаратов, диуретиков, переливание плазмы.
В уремической стадии болезни может понадобиться хронический почечный диализ или даже трансплантация почки. Для пациентов, у которых диагностирован амилоидоз почек, лечение требует значительного изменения пищевого рациона: ограничение соли и белка, повышенное употребление углеводов, а также пищи, богатой солями камня и витаминами.
Данная статья размещена исключительно с целью ознакомления в познавательных целях и не является научным материалом или профессиональным медицинским советом. За диагностикой и лечением обратитесь к врачу.
Амилоидоз почек
Амилоидоз почек (АП) — заболевание с внеклеточным отложением в ткани почек амилоида, развивающееся преимущественно при генерализованных (системных) формах амилоидоза.
Патоморфология
Отложения амилоида в органах и тканях .Почки увеличены, светло-жёлтые с гладкой поверхностью — «большая жировая почка» . Амилоид откладывается преимущественно в клубочках . Распространённость отложений амилоида в почке нарастает по мере прогрессирования амилоидоза . После окрашивания конго красным при поляризационной микроскопии амилоид обнаруживают по зелёному цвету . Электронная микроскопия позволяет установить окончательный диагноз.
Клиничвская картина
Характерно сочетание признаков поражения почек и внепочечных проявлений, что создаёт полиморфную клиническую картину
Клинические проявления амилоидоза почек в зависимости от стадии. Выделяют четыре стадии амилоидоза почек :
Латентная (отложения амилоида в почках есть, однако клинические проявления и протеинурия отсутствуют).
Протеинурическая — протеинурия сначала минимальная, затем нарастает. Микрогематурия (3–11,5%), асептическая лейкоцитурия (до 40%). Возможно повышение СОЭ .
Нефротическая -проявления нефротического синдрома — сочетание массивной протеинурии, гипопротеинемии, гиперхолестеринемии и отёков, резистентных к диуретикам .Высокая СОЭ.Артериальная гипертензия (редко при АL-амилоидозе, до 23% при АА-амилоидозе — на стадии ХПН) . По УЗИ — увеличенные уплотнённые почки •
Азотемическая (ХПН) -отёки обычно сохраняются в терминальной стадии .Артериальная гипертензия чаще невысокая.
Лечение
Общая тактика
Режим амбулаторный, за исключением тяжёлых состояний (выраженная сердечная недостаточность, ХПН) . Больным амилоидозом показан длительный (1,5–2 года) приём сырой печени (по 100–120 г/сут) .Ограничение потребления белка .Ограничение потребления соли при ХПН и сердечной недостаточности . При вторичном амилоидозе — лечение основного заболевания (туберкулёз, остеомиелит, эмпиема плевры и др.), после радикального устранения нередко исчезают симптомы амилоидоза .Перевод пациента с диализным амилоидозом на перитонеальный диализ, удаляющий (в отличие от обычного гемодиализа) 2-микроглобулин .При амилоидозе кишечника, протекающем с упорной диареей, — вяжущие средства (нитрат висмута основного, адсорбенты) . Лечение нефротического синдрома . В терминальной стадии ХПН гемодиализ, возможна трансплантация почки.
Лекарственная терапия
При первичном амилоидозе .В начальных стадиях процесса — хлорохин в дозе 0,25 г 1 р/сут длительно . Сочетание мелфалана 0,15 мг/кг и преднизолон по 0,8 мг/кг 7-дневными курсами с перерывом 4-–6 нед в течение 6 мес–1 года .Сочетание мелфалана, преднизолона и колхицина . Полихимиотерапия с включением винкристина, доксорубицина, циклофосфамид, мелфалана, дексаметазона в различных комбинациях с последующей трансплантацией стволовых клеток .При вторичном амилоидозе .Специфическое лечение основного заболевания . Такрин — при деменции слабой или средней степени (болезнь Альцхаймера) . При семейном амилоидозе — колхицин (по 0,6 мг 2–3 р/сут) . Лечение сердечной недостаточности массивными дозами мочегонных (противопоказаны сердечные гликозиды, блокаторы кальциевых каналов, b-адреноблокаторы) . Симптоматическая терапия: витамины, диуретики, антигипертензивные средства, переливание плазмы и т.д.
Хирургическое лечение
Спленэктомия позволяет уменьшить количество амилоида, образующегося в организме . При изолированном опухолевидном амилоидозе ЖКТ — оперативное лечение . Пересадка сердца (в стадии внедрения) . Пересадка печени при АТТR-амилоидозе.
Течение и прогноз
Первичный амилоидоз . После развития ХПН пациенты обычно живут менее года . После развития сердечной недостаточности пациенты обычно живут около 4 мес . Средняя выживаемость — 12–14 мес . Амилоидоз, ассоциированный с миеломой, имеет наименее благоприятный прогноз . Вторичный амилоидоз: прогноз определяется возможностью лечения основного заболевания . Семейный и диализный амилоидоз: прогноз очень вариабелен, заболевание протекает тяжелее у лиц пожилого возраста.
Амилоидоз и заболевание почек | NIDDK
Что такое амилоидоз?
Амилоидоз — редкое заболевание, которое возникает, когда амилоидные белки откладываются в тканях и органах. Амилоидные белки — это аномальные белки, которые организм не может расщепить и переработать, как это происходит с нормальными белками. Когда амилоидные белки слипаются, они образуют амилоидные отложения. Накопление этих отложений повреждает органы и ткани человека. Амилоидоз может поражать разные органы и ткани у разных людей и может поражать более одного органа одновременно.Амилоидоз чаще всего поражает почки, сердце, нервную систему, печень и пищеварительный тракт. Симптомы и тяжесть амилоидоза зависят от пораженных органов и тканей.
Что такое почки и что они делают?
Почки — это два органа в форме бобов, каждый размером с кулак. Они расположены чуть ниже грудной клетки, по одной с каждой стороны позвоночника. Каждый день две почки фильтруют от 120 до 150 литров крови, чтобы произвести от 1 до 2 литров мочи, состоящей из отходов и лишней жидкости.Моча течет из почек в мочевой пузырь по трубкам, называемым мочеточниками. Мочевой пузырь хранит мочу. Когда мочевой пузырь опорожняется, моча выходит из организма через трубку, называемую уретрой, расположенную в нижней части мочевого пузыря. У мужчин уретра длинная, у женщин — короткая.
Почки — это два органа в форме бобов, каждый размером с кулак.Какие типы амилоидоза поражают почки?
Первичный амилоидоз и амилоидоз, связанный с диализом, — это типы амилоидоза, которые могут поражать почки.
Первичный амилоидоз почек
Почки — это органы, наиболее часто поражаемые первичным амилоидозом. Амилоидные отложения повреждают почки и затрудняют им фильтрацию отходов и расщепление белков. Когда почки становятся слишком поврежденными, они больше не могут функционировать достаточно хорошо для поддержания здоровья, что приводит к почечной недостаточности. Почечная недостаточность может привести к таким проблемам, как высокое кровяное давление, болезни костей и анемия — состояние, при котором в организме меньше эритроцитов, чем обычно.
Амилоидоз, связанный с диализом
У людей, страдающих почечной недостаточностью и находящихся на длительном диализе, может развиться амилоидоз, связанный с диализом. Этот тип амилоидоза возникает, когда в крови накапливается определенный белок, называемый микроглобулином бета-2, поскольку диализ не удаляет его полностью. Два типа диализа —
- гемодиализ. Гемодиализ использует специальный фильтр, называемый диализатором, для удаления отходов и лишней жидкости из крови.
- перитонеальный диализ. При перитонеальном диализе для фильтрации крови используется слизистая оболочка брюшной полости — пространство в теле, в котором находятся такие органы, как желудок, кишечник и печень.
Амилоидоз, связанный с диализом, является осложнением почечной недостаточности, поскольку ни гемодиализ, ни перитонеальный диализ не эффективно фильтруют бета-2-микроглобулин из крови. В результате в крови остается повышенное количество бета-2-микроглобулина.
Каковы признаки и симптомы первичного амилоидоза почек?
Наиболее частым признаком первичного амилоидоза почек является нефротический синдром — совокупность признаков, указывающих на поражение почек.Признаки нефротического синдрома включают
- альбуминурия — повышенное количество белка, альбумина, в моче. Человек с нефротическим синдромом выделяет более половины чайной ложки альбумина в день.
- гиперлипидемия — состояние, при котором в крови человека содержится больше, чем обычно, жиров и холестерина.
- отек — опухоль, обычно в ногах, ступнях или лодыжках человека, реже — в руках или лице.
- гипоальбуминемия — состояние, при котором в крови человека содержится меньше, чем обычно, альбумина.
Другие признаки и симптомы первичного амилоидоза могут включать
- Усталость или чувство усталости
- одышка
- низкое кровяное давление
- Онемение, покалывание или жжение в руках или ногах
- потеря веса
Каковы симптомы диализного амилоидоза?
Симптомы диализного амилоидоза могут включать
- Боль, скованность и жидкость в суставах.
- аномальные, содержащие жидкость мешочки, называемые кистами, в некоторых костях.
- синдром запястного канала, вызванный необычным накоплением амилоидных белков в запястьях. Симптомы синдрома запястного канала включают онемение или покалывание, иногда связанное с мышечной слабостью, в пальцах и руках.
Связанный с диализом амилоидоз чаще всего поражает кости, суставы и ткани, соединяющие мышцы с костью, называемые сухожилиями. Заболевание также может поражать пищеварительный тракт и такие органы, как сердце и легкие.Костные кисты, вызванные диализным амилоидозом, могут привести к переломам костей. Связанный с диализом амилоидоз также может вызывать разрывы сухожилий и связок. Связки — это ткани, которые соединяют кости с другими костями.
Как диагностируется первичный амилоидоз почек?
Врач диагностирует первичный амилоидоз почек с помощью
- Медицинский и семейный анамнез
- медицинский осмотр
- общий анализ мочи
- Анализы крови
- биопсия почки
Медицинский и семейный анамнез
Медицинский и семейный анамнез может помочь врачу диагностировать амилоидоз почек.Он или она попросят пациента предоставить медицинский и семейный анамнез.
Физический осмотр
Медицинский осмотр может помочь диагностировать первичный амилоидоз почек. Во время медицинского осмотра врач обычно
- исследует тело пациента на опухоль
- использует стетоскоп для прослушивания легких
- постукивает по определенным участкам тела пациента
Анализ мочи
Медицинский работник может использовать анализ мочи — анализ образца мочи — для проверки содержания альбумина и амилоидных белков в моче.Пациент сдает образец мочи в специальном контейнере в офисе врача или в коммерческом учреждении. Медсестра или технический специалист могут протестировать образец в том же месте или отправить его в лабораторию для анализа. Содержание альбумина в моче, превышающее норму, может указывать на повреждение почек из-за первичного амилоидоза. Амилоидные белки в моче могут указывать на амилоидоз.
Анализы крови
Врач может использовать анализы крови, чтобы увидеть, насколько хорошо работают почки, и проверить наличие амилоидных белков и гиперлипидемии.Анализ крови включает в себя забор крови пациента в офисе поставщика медицинских услуг или в коммерческом учреждении и отправку образца в лабораторию для анализа. Анализы крови на функцию почек определяют количество продуктов жизнедеятельности в крови, которые обычно отфильтровываются здоровыми почками. Гиперлипидемия может указывать на нефротический синдром. Амилоидные белки в крови могут указывать на амилоидоз.
Биопсия почки
Только биопсия может показать отложения амилоидного белка в почках. Врач может порекомендовать биопсию почки, если другие тесты показывают повреждение почек.Биопсия почки — это процедура, которая включает взятие кусочка ткани почек для исследования под микроскопом. Врач выполняет биопсию почки в больнице с легкой седацией и местной анестезией. Поставщик медицинских услуг использует методы визуализации, такие как ультразвук или компьютерная томография (КТ), чтобы ввести иглу для биопсии в почку и взять образец ткани. Патолог — врач, специализирующийся на диагностике заболеваний — исследует ткань в лаборатории на наличие амилоидных белков и повреждений почек.
Результаты биопсии могут помочь врачу определить лучший курс лечения.
Как диагностируется амилоидоз, связанный с диализом?
Поставщик медицинских услуг диагностирует связанный с диализом амилоидоз с помощью
- Общий анализ мочи
- Анализы крови
- визуализационные тесты
Медицинский работник может использовать общий анализ мочи и анализы крови для определения количества амилоидных белков в моче и крови. Визуализирующие обследования, такие как рентген и компьютерная томография, могут предоставить изображения костных кист и амилоидных отложений в костях, суставах, сухожилиях и связках.Техник-рентгенолог выполняет визуализационные тесты в офисе поставщика медицинских услуг, амбулаторном центре или больнице. Радиолог — врач, специализирующийся на медицинской визуализации — интерпретирует изображения. Пациенту не требуется анестезия.
Рентгеновский снимок, показывающий отложения амилоида на запястьеКак лечится первичный амилоидоз почек?
Медицинский работник лечит первичный амилоидоз почек следующими препаратами:
- Медикаментозная терапия, в том числе химиотерапия
- трансплантат стволовых клеток
- Лечение других заболеваний
Медикаментозная терапия. Целью медикаментозной терапии, включая химиотерапию, является снижение уровня амилоидного белка в крови. Многие поставщики медицинских услуг рекомендуют комбинированную медикаментозную терапию, например
.- мелфалан (Алькеран), вид химиотерапии
- дексаметазон (декадрон), противовоспалительный стероидный препарат
Эти лекарства могут остановить рост клеток, вырабатывающих амилоидные белки. Эти лекарства могут вызвать выпадение волос и серьезные побочные эффекты, такие как тошнота, рвота и усталость.
Пересадка стволовых клеток. Трансплантация стволовых клеток — это процедура, при которой поврежденные стволовые клетки пациента заменяются здоровыми. Стволовые клетки находятся в костном мозге и превращаются в три типа клеток крови, в которых нуждается организм. Чтобы подготовиться к трансплантации стволовых клеток, пациент получает высокие дозы химиотерапии. Фактическая трансплантация похожа на переливание крови. Пересаженные стволовые клетки попадают в костный мозг, чтобы произвести новые здоровые клетки крови. Химиотерапия, которую пациент получает для подготовки к трансплантации, может иметь серьезные побочные эффекты, поэтому важно поговорить с врачом о рисках этой процедуры.
Подробнее см. Трансплантация крови и костного мозга.
Лечение других заболеваний. Первичный амилоидоз неизлечим, поэтому лечение некоторых побочных эффектов и других состояний, наблюдаемых при этом заболевании, имеет важное значение. Другие условия могут включать
- анемия — лечение может включать лекарства
- Депрессия — лечение может включать беседу с психологом и прием лекарств
- Усталость — лечение может включать изменение диеты и уровня активности
- Заболевание почек — лечение может включать лекарства, помогающие поддерживать функцию почек или замедлять прогрессирование заболевания почек.
Пациенту и его семье следует обсудить с лечащим врачом ресурсы для поддержки и вариантов лечения.
Как лечится диализный амилоидоз?
Поставщик медицинских услуг лечит связанный с диализом амилоидоз с помощью
- медикаментозная терапия
- новые и эффективные фильтры для гемодиализа
- хирургия
- трансплантат почки
Целью медикаментозной терапии и использования новых, более эффективных фильтров для гемодиализа является снижение уровня амилоидного белка в крови. Медикаментозная терапия может помочь уменьшить такие симптомы, как боль и воспаление.Поставщик медицинских услуг может лечить человека с диализным амилоидозом, у которого есть проблемы с костями, суставами и сухожилиями, такие как костные кисты и синдром запястного канала, с помощью хирургического вмешательства.
Амилоидоз, связанный с диализом, неизлечим; однако успешная пересадка почки может остановить прогрессирование болезни.
Питание, диета и питание
Исследователи не обнаружили, что еда, диета и питание играют роль в возникновении или предотвращении первичного амилоидоза почек или амилоидоза, связанного с диализом.Люди с нефротическим синдромом могут вносить изменения в рацион питания, например,
- ограничение натрия в пище (PDF, 167 КБ) , часто из соли, для уменьшения отеков и снижения артериального давления
- уменьшение потребления жидкости для уменьшения отеков и снижения артериального давления
- придерживаться диеты с низким содержанием насыщенных жиров и холестерина, чтобы контролировать количество жиров и холестерина в крови, превышающее норму
Медицинские работники могут рекомендовать людям с заболеванием почек употреблять умеренное или пониженное количество белка.Белки распадаются на продукты жизнедеятельности, которые почки фильтруют из крови. Употребление в пищу большего количества белка, чем необходимо организму, может обременить почки и привести к более быстрому ухудшению функции почек. Однако слишком низкое потребление белка может привести к недоеданию — состоянию, которое возникает, когда организм не получает достаточного количества питательных веществ.
Люди с заболеваниями почек, соблюдающие диету с ограничением белков (PDF, 112 КБ) должны сдавать анализы крови, которые могут показать низкий уровень питательных веществ. Людям с первичным амилоидозом почек или амилоидозом, связанным с диализом, следует поговорить с врачом об ограничениях в питании, чтобы наилучшим образом удовлетворить свои индивидуальные потребности.
Если вы узнаете о своем лечении как можно больше, это поможет вам стать важным членом вашей медицинской бригады.
Клинические испытания
Национальный институт диабета, болезней органов пищеварения и почек (NIDDK) и другие подразделения Национального института здоровья (NIH) проводят и поддерживают исследования многих заболеваний и состояний.
Что такое клинические испытания и подходят ли они вам?
Клинические испытания являются частью клинических исследований и лежат в основе всех достижений медицины.Клинические испытания ищут новые способы предотвращения, обнаружения или лечения заболеваний. Исследователи также используют клинические испытания для изучения других аспектов лечения, таких как улучшение качества жизни людей с хроническими заболеваниями. Узнайте, подходят ли вам клинические испытания.
Какие клинические испытания открыты?
Клинические испытания, которые в настоящее время открыты и набираются, можно просмотреть на сайте www.ClinicalTrials.gov.
Причины, типы, симптомы, диагностика, лечение и прогноз
Амилоидоз — это когда аномальный белок, называемый амилоидом, накапливается в ваших тканях и органах.Когда это происходит, это влияет на их форму и то, как они работают. Амилоидоз — серьезная проблема для здоровья, которая может привести к опасной для жизни органной недостаточности.
Причины и типы амилоидоза
Многие различные белки могут приводить к отложению амилоида, но лишь некоторые из них связаны с серьезными проблемами со здоровьем. Тип белка и место его скопления говорят о типе амилоидоза. Амилоидные отложения могут накапливаться по всему телу или только в одной области.
Различные типы амилоидоза включают:
AL амилоидоз (амилоидоз легкой цепи иммуноглобулина). Это наиболее распространенный тип, который раньше назывался первичным амилоидозом. AL означает «легкие амилоидные цепи», тип белка, ответственного за это состояние. Причины неизвестны, но это случается, когда костный мозг вырабатывает аномальные антитела, которые невозможно расщепить. Это связано с раком крови, называемым множественной миеломой. Это может повлиять на ваши почки, сердце, печень, кишечник и нервы.
Продолжение
АА амилоидоз. Ранее известное как вторичный амилоидоз, это состояние является результатом другого хронического инфекционного или воспалительного заболевания, такого как ревматоидный артрит, болезнь Крона или язвенный колит.В основном это влияет на ваши почки, но также может повлиять на пищеварительный тракт, печень и сердце. AA означает, что белок амилоидного типа A вызывает этот тип.
Амилоидоз, связанный с диализом (DRA). Это чаще встречается у пожилых людей и людей, находящихся на диализе более 5 лет. Эта форма амилоидоза вызвана отложениями бета-2-микроглобулина, которые накапливаются в крови. Отложения могут накапливаться во многих различных тканях, но чаще всего поражают кости, суставы и сухожилия.
Семейный или наследственный амилоидоз. Это редкая форма, передающаяся от семьи. Часто поражает печень, нервы, сердце и почки. Многие генетические дефекты связаны с более высокой вероятностью амилоидного заболевания. Например, причиной может быть аномальный белок, такой как транстиретин (TTR).
Возрастной (старческий) системный амилоидоз. Это вызвано отложениями нормального TTR в сердце и других тканях. Чаще всего это случается у пожилых мужчин.
Органоспецифический амилоидоз. Это вызывает отложения амилоидного белка в отдельных органах, включая кожу (кожный амилоидоз).
Хотя некоторые типы отложений амилоида связаны с болезнью Альцгеймера, мозг редко поражается амилоидозом, который встречается повсюду в организме.
Факторы риска амилоидоза
Мужчины заболевают амилоидозом чаще, чем женщины. Риск амилоидоза повышается с возрастом. Амилоидоз поражает 15% пациентов с формой рака, называемой множественной миеломой.
Амилоидоз также может развиться у людей с терминальной стадией заболевания почек, которые длительное время находятся на диализе (см. «Амилоидоз, связанный с диализом» выше).
Симптомы амилоидоза
Симптомы амилоидоза часто незаметны. Они также могут сильно различаться в зависимости от того, где в организме накапливается амилоидный белок. Важно отметить, что описанные ниже симптомы могут быть вызваны различными проблемами со здоровьем. Только ваш врач может поставить диагноз амилоидоза.
Общие симптомы амилоидоза могут включать:
- Изменение цвета кожи
- Сильная усталость
- Ощущение полноты
- Боль в суставах
- Низкое количество эритроцитов (анемия)
- Одышка
- Отек языка
- Покалывание и онемение в ногах и стопах
- Слабая хватка
- Сильная слабость
- Внезапная потеря веса
Сердечный (сердечный) амилоидоз
Отложения амилоида в сердце могут сделать стенки сердечной мышцы жесткими.Они также могут ослабить сердечную мышцу и повлиять на электрический ритм сердца. Это состояние может привести к уменьшению притока крови к сердцу. В конце концов, ваше сердце больше не сможет нормально перекачивать кровь. Если амилоидоз поражает ваше сердце, у вас может быть:
- Одышка с легкой активностью
- Нерегулярное сердцебиение
- Признаки сердечной недостаточности, включая отек стоп и лодыжек, слабость, утомляемость и тошноту, среди прочего
Амилоидоз почек (почек)
Почки фильтруют отходы и токсины из крови.Амилоидные отложения в почках мешают им выполнять эту работу. Когда ваши почки не работают должным образом, в вашем теле накапливаются вода и опасные токсины. Если амилоидоз поражает почки, у вас могут быть:
- Признаки почечной недостаточности, включая отек стоп и лодыжек и отечность вокруг глаз
- Высокий уровень белка в моче
Желудочно-кишечный амилоидоз
Амилоидные отложения вдоль вашего тела. Желудочно-кишечный тракт замедляет движение пищи через кишечник.Это мешает пищеварению. Если амилоидоз поражает ваш желудочно-кишечный тракт, у вас может быть:
- Снижение аппетита
- Диарея
- Тошнота
- Боль в желудке
- Потеря веса
Если ваша печень поражена, это может вызвать ее увеличение и скопление жидкости в организме .
Амилоидная нейропатия
Отложения амилоида могут повредить нервы вне головного и спинного мозга, которые называются периферическими нервами. Периферические нервы несут информацию между головным и спинным мозгом и остальным телом.Например, они заставляют ваш мозг воспринимать боль, если вы обожжете руку или ударите пальцы ног. Если амилоидоз поражает нервы, у вас могут быть:
- Проблемы с равновесием
- Проблемы с контролем мочевого пузыря и кишечника
- Проблемы с потоотделением
- Покалывание и слабость
- Головокружение при стоянии из-за нарушения способности вашего тела контролировать кровяное давление
Диагностика амилоидоза
Тщательное медицинское обследование и подробное и точное описание вашей истории болезни имеют решающее значение для того, чтобы помочь вашему врачу диагностировать амилоидоз.
Анализы крови и мочи могут выявить аномальные белки. В зависимости от ваших симптомов врач также может проверить вашу щитовидную железу и печень.
Ваш врач проведет биопсию, чтобы подтвердить диагноз амилоидоза и узнать, какой у вас конкретный тип белка. Образец ткани для биопсии может быть взят из жира на животе (брюшной жировой подушечки), костного мозга или иногда изо рта, прямой кишки или других органов. Не всегда необходимо проводить биопсию части тела, поврежденной амилоидными отложениями.
Также могут помочь визуализирующие тесты. Они показывают степень повреждения таких органов, как сердце, печень или селезенка.
Ваш врач проведет генетический тест, если он сочтет, что у вас тип, передающийся от семьи. Лечение наследственного амилоидоза отличается от лечения других типов заболевания.
После постановки диагноза врач может проверить ваше сердце с помощью эхокардиограммы или вашу печень и селезенку с помощью визуализационных тестов.
Лечение амилоидоза
Лекарства от амилоидоза нет.Ваш врач назначит лечение, чтобы замедлить выработку амилоидного белка и облегчить симптомы. Если амилоидоз связан с другим заболеванием, лечение будет включать в себя лечение этого основного состояния.
Конкретное лечение зависит от типа амилоидоза и количества пораженных органов.
- Высокодозная химиотерапия с трансплантацией стволовых клеток может помочь удалить вещество, которое приводит к образованию амилоида у некоторых людей с первичным амилоидозом AL.Только химиотерапевтические препараты могут использоваться для лечения других пациентов с первичным амилоидозом AL.
- Вторичный амилоидоз (AA) лечится путем контролирования основного заболевания и с помощью мощных противовоспалительных препаратов, называемых стероидами, которые борются с воспалением.
- Пересадка печени может помочь в лечении этого заболевания, если у вас есть определенные типы наследственного амилоидоза.
- Новые методы лечения могут замедлить выработку аномального белка TTR.
- Ваш врач может также порекомендовать пересадку почки.
Другие методы лечения симптомов включают:
- Мочегонные препараты для удаления лишней воды из вашего тела
- Загустители для добавления в жидкости, чтобы предотвратить удушье, если у вас есть проблемы с глотанием
- Компрессионные чулки для снятия отеков в ногах или ступнях
- Изменения в том, что вы едите, особенно если у вас желудочно-кишечный амилоидоз
Чего ожидать
Амилоидоз может быть смертельным, особенно если он поражает ваше сердце или почки.Ранняя диагностика и лечение важны и могут помочь улучшить выживаемость.
Исследователи продолжают задаваться вопросом, почему некоторые типы амилоида вызывают у людей заболевание и как можно остановить образование амилоида. Исследования по поиску новых методов лечения продолжаются. Если у вас амилоидоз, подумайте о том, чтобы спросить своего врача, есть ли какие-либо клинические испытания, к которым вы можете присоединиться, или найдите их, посетив www.clinicaltrials.gov и введя поисковый запрос «амилоидоз».
Амилоидоз — Диагностика и лечение
Диагноз
Амилоидоз часто упускают из виду, потому что признаки и симптомы могут имитировать симптомы более распространенных заболеваний.
Ранняя диагностика может помочь предотвратить дальнейшее повреждение органов. Точный диагноз важен, потому что лечение сильно различается в зависимости от вашего конкретного состояния.
Лабораторные исследования
Ваша кровь и моча могут быть проанализированы на аномальный белок, который может указывать на амилоидоз. В зависимости от ваших признаков и симптомов вам также могут пройти функциональные пробы щитовидной железы и печени.
Биопсия
Можно взять образец ткани и проверить на наличие признаков амилоидоза.Биопсия может быть взята из подкожного жира на животе (аспират жира), костного мозга или пораженного органа, например печени или почки. Специализированное исследование ткани может помочь определить тип амилоидного отложения.
Визуальные тесты
Изображения органов, пораженных амилоидозом, могут помочь определить степень вашего заболевания. Тесты могут включать:
- Эхокардиограмма. Эта технология использует звуковые волны для создания движущихся изображений, которые могут показать, насколько хорошо работает ваше сердце.Он также может указывать на поражение сердца, которое может быть специфическим для определенных типов амилоидоза.
- Магнитно-резонансная томография (МРТ). MRI использует радиоволны и сильное магнитное поле для создания детальных изображений органов и тканей вашего тела. Их можно использовать для оценки структуры и функции вашего сердца.
- Ядерная визуализация. В этом тесте крошечные количества радиоактивного материала (индикаторы) вводятся в вену. Это может выявить раннее повреждение сердца, вызванное определенными типами амилоидоза.Это также может помочь различать разные типы амилоидоза, что может помочь в принятии решения о лечении.
Лечение
Лекарства от амилоидоза нет. Но лечение может помочь справиться с признаками и симптомами и ограничить дальнейшее производство амилоидного белка. Если амилоидоз был вызван другим заболеванием, например ревматоидным артритом или туберкулезом, лечение основного заболевания может оказаться полезным.
Лекарства
- Химиотерапия. Многие из тех же типов лекарств, которые используются для лечения некоторых форм рака, используются при амилоидозе AL, чтобы остановить рост аномальных клеток, которые производят белок, ведущий к образованию амилоида.
- Сердечные препараты. Если ваше сердце поражено, ваш врач может порекомендовать препараты, разжижающие кровь, чтобы снизить риск образования тромбов, и лекарства, контролирующие частоту сердечных сокращений. Вам также может потребоваться ограничить потребление соли и принять лекарства, усиливающие мочеиспускание, что может снизить нагрузку на сердце и почки.
- Таргетная терапия. При некоторых типах амилоидоза такие препараты, как патисиран (Onpattro) и инотерсен (Тегседи), могут мешать командам, посылаемым ошибочными генами, создающими амилоид. Другие препараты, такие как тафамидис (Vyndamax, Vyndaqel) и дифлунисал, могут стабилизировать кусочки белка в кровотоке и предотвратить их превращение в амилоидные отложения.
Хирургические и другие процедуры
- Трансплантация аутологичных стволовых клеток крови. Эта процедура включает в себя сбор ваших собственных стволовых клеток из крови через вену и их кратковременное хранение, пока вы проходите высокодозную химиотерапию. Затем стволовые клетки возвращаются в ваше тело через вену. Это лечение наиболее подходит для людей, у которых болезнь не запущена и сердце не сильно пострадало.
- Диализ. Если ваши почки были повреждены амилоидозом, вам может потребоваться начать диализ. В этой процедуре используется машина для регулярной фильтрации шлаков, солей и жидкости из вашей крови.
- Пересадка органов. Ваш врач может предложить операцию по замене сердца или почек, если отложения амилоида серьезно повредили эти органы. Некоторые типы амилоида образуются в печени, поэтому трансплантация печени может остановить это производство.
Клинические испытания
Изучите исследования клиники Mayo Clinic, в которых тестируются новые методы лечения, вмешательства и тесты как средства предотвращения, обнаружения, лечения или контроля этого состояния.
Подготовка к приему
Вас могут направить к врачу, специализирующемуся на заболеваниях крови (гематологу).
Что вы можете сделать
- Запишите свои симптомы, включая те, которые могут показаться не связанными с причиной, по которой вы записались на прием.
- Составьте список всех ваших лекарств, витаминов и пищевых добавок.
- Запишите основную медицинскую информацию, включая другие состояния.
- Запишите ключевую личную информацию, включая любые недавние изменения или факторы стресса в вашей жизни.
- Запишите вопросы, которые задайте своему врачу.
- Попросите родственника или друга сопровождать вас, , чтобы помочь вам вспомнить, что говорит врач.
Вопросы, которые следует задать врачу
- Какая наиболее вероятная причина моих симптомов?
- Какой у меня амилоидоз?
- Какие органы поражены?
- На какой стадии мое заболевание?
- Какие тесты мне нужны?
- Какое лечение мне нужно?
- Есть ли у меня риск долгосрочных осложнений?
- Каких побочных эффектов можно ожидать от лечения?
- Нужно ли мне соблюдать какие-либо ограничения в питании или активности?
- У меня другое заболевание.Как мне лучше всего управлять ими вместе?
В дополнение к вопросам, которые вы подготовили задать своему врачу, не стесняйтесь задавать другие вопросы во время приема.
Чего ожидать от врача
Ваш врач, скорее всего, задаст вам ряд вопросов. Готовность ответить на них может дать время, чтобы обсудить те моменты, на которые вы хотите потратить больше времени. Вас могут спросить:
- Когда у вас впервые появились симптомы? Насколько они серьезны, являются ли они постоянными или случайными?
- Кажется, что-нибудь улучшает или ухудшает ваши симптомы?
- Как твой аппетит? Вы недавно похудели, даже не пытаясь?
- Были ли у вас отеки на ногах?
- Испытывали ли вы одышку?
- Можете ли вы работать и выполнять обычные повседневные задачи? Вы часто устаете?
- Вы заметили, что у вас легко появляются синяки?
- У кого-нибудь в вашей семье когда-либо диагностировали амилоидоз?
Лечение амилоидоза в клинике Мэйо
14 марта 2020 г.
Показать ссылки- Goldman L, et al., ред. Амилоидоз. В: Медицина Гольдмана-Сесила. 26-е изд. Эльзевир; 2020. https://www.clinicalkey.com. По состоянию на 1 октября 2019 г.
- Hoffman R, et al. Амилоидоз легкой цепи иммуноглобулина (первичный амилоидоз). В кн .: Гематология: основные принципы и практика. 7-е изд. Эльзевир; 2018. https://www.clinicalkey.com. По состоянию на 1 октября 2019 г.
- Амилоидоз. Руководство Merck Professional Version. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/amyloidosis/amyloidosis?query=amyloidosis.По состоянию на 1 октября 2019 г.
- Ferri FF. Амилоидоз. В: Клинический советник Ферри 2020. Elsevier; 2020. https://www.clinicalkey.com. По состоянию на 1 октября 2019 г.
- Gorevic PD. Обзор амилоидоза. https://www.uptodate.com/contents/search. По состоянию на 1 октября 2019 г.
- AskMayoExpert. Сердечный амилоидоз. Клиника Майо; 2018.
- Fontana M, et al. Амилоидоз миокарда: типичное интерстициальное заболевание. Журнал Американского колледжа кардиологии: сердечно-сосудистые изображения.2019. doi: 10.1016 / j.jcmg.2019.06.023.
- AskMayoExpert. Амилоидоз почек. Клиника Майо; 2019.
- Feehally J, et al. Амилоидоз почек и заболевания клубочков с отложением моноклональных иммуноглобулинов. В кн .: Комплексная клиническая нефрология. 6-е изд. Эльзевир. 2019. https://www.clinicalkey.com. По состоянию на 1 октября 2019 г.
- Morrow ES Jr. Allscripts EPSi. Клиника Майо. 1 ноября 2019 г.
- Прути РК (заключение эксперта). Клиника Майо. 13 января 2020 г.
Связанные
Продукты и услуги
Показать больше продуктов и услуг Mayo ClinicМедицинская помощь, хирургическая помощь, консультации
Bustamante JG, Brito D.Амилоидоз. 2018, январь [Medline]. [Полный текст].
Ostertag B. Демонстрация собственного семейства параамилоидоза. Zentralbl Aug Pathol . 1932. 56: 253-4.
Benson MD, Liepnieks J, Uemichi T, et al. Наследственный амилоидоз почек, связанный с мутантной альфа-цепью фибриногена. Нат Генет . 1993 3 марта (3): 252-5. [Медлайн].
Booth DR, Tan SY, Booth SE и др. Новый вариант аполипопротеина Al, Trp50Arg, вызывает наследственный амилоидоз. QJM . 1995 Октябрь 88 (10): 695-702. [Медлайн].
Booth DR, Tan SY, Booth SE и др. Наследственный печеночный и системный амилоидоз, вызванный новой мутацией делеции / вставки в гене аполипопротеина AI. Дж. Клин Инвест . 15 июня 1996 г., 97 (12): 2714-21. [Медлайн].
de Sousa MM, Vital C, Ostler D, et al. Аполипопротеин AI и транстиретин как компоненты амилоидных фибрилл в родстве с амилоидозом apoAI Leu178His. Ам Дж. Патол . 2000 июн. 156 (6): 1911-7. [Медлайн].
Гиллмор Дж. Д., Бут Д. Р., Мадху С. и др. Наследственный амилоидоз почек, связанный с вариантным лизоцимом в большой английской семье. Циферблат нефрола . 1999 14 ноября (11): 2639-44. [Медлайн].
Hamidi Asl K, Liepnieks JJ, Nakamura M, et al. Новый вариант аполипопротеина A-1, Arg173Pro, ассоциированный с сердечным и кожным амилоидозом. Биохимия Биофиз Рес Коммуна .1999 13 апреля. 257 (2): 584-8. [Медлайн].
Hamidi Asl L, Fournier V, Billerey C, et al. Мутация альфа-цепи фибриногена (Arg554 Leu), связанная с наследственным амилоидозом почек во французской семье. Амилоид . 1998 Декабрь 5 (4): 279-84. [Медлайн].
Hamidi Asl L, Liepnieks JJ, Hamidi Asl K, et al. Наследственная амилоидная кардиомиопатия, вызванная вариантом аполипопротеина А1. Ам Дж. Патол . 1999, январь, 154 (1): 221-7. [Медлайн].
Hamidi Asl L, Liepnieks JJ, Uemichi T, et al. Почечный амилоидоз с мутацией сдвига рамки считывания в гене альфа-цепи фибриногена, продуцирующий новый амилоидный белок. Кровь . 1997 15 декабря. 90 (12): 4799-805. [Медлайн].
Джонс Л.А., Хардинг Дж. А., Коэн А.С.. Новое семейство США имеет вариант аполипопротеина AI (Arg26). Амилоид и амилоидоз . 1991. 385-8.
Obici L, Bellotti V, Mangione P и др.Новый вариант аполипопротеина A-I leu (174) -> Ser вызывает наследственный кардиальный амилоидоз, а амилоидные фибриллы состоят из 93 остатков N-концевого полипептида. Ам Дж. Патол . 1999 Сентябрь 155 (3): 695-702. [Медлайн].
Persey MR, стенд DR, стенд SE. Новая делеционная мутация гена аполипопротеина AI, вызывающая наследственный амилоидоз. Clin Sci . 1996. 90 (Дополнение 34): 33.
Persey MR, Booth DR, Booth SE, et al.Наследственный нефропатический системный амилоидоз, вызванный новым вариантом аполипопротеина A-I. Почки Инт . 1998 Февраль 53 (2): 276-81. [Медлайн].
Сутар А.К., Хокинс П.Н., Вигушин Д.М. и др. Мутация аполипопротеина AI Arg-60 вызывает аутосомно-доминантный амилоидоз. Proc Natl Acad Sci U S A . 1992 15 августа. 89 (16): 7389-93. [Медлайн].
Uemichi T, Liepnieks JJ, Alexander F, Benson MD. Молекулярные основы амилоидоза почек в ирландско-американских и польско-канадских родословных. QJM . 1996 Октябрь 89 (10): 745-50. [Медлайн].
Uemichi T, Liepnieks JJ, Benson MD. Наследственный амилоидоз почек с новым вариантом фибриногена. Дж. Клин Инвест . 1994 Февраль 93 (2): 731-6. [Медлайн].
Uemichi T, Liepnieks JJ, Gertz MA, Benson MD. Альфа-цепь фибриногена А Leu 554: афроамериканец с поздним началом амилоидоза почек. Амилоид . 1998 Сентябрь 5 (3): 188-92. [Медлайн].
Уэмичи Т., Лиепниекс Дж. Дж., Ямада Т. и др.Мутация сдвига рамки считывания в гене альфа-цепи фибриногена А в родстве с амилоидозом почек. Кровь . 1996 15 мая. 87 (10): 4197-203. [Медлайн].
Li Z, Xu H, Liu D, Li D, Liu G, Wang SX. Наследственный амилоидоз почек с вариантом лизоцима p.Trp82Arg в китайской семье: отчет о болезни и обзор литературы. БМК Нефрол . 2019 8 августа. 20 (1): 310. [Медлайн]. [Полный текст].
Хокинс П.Н., Майерс М.Дж., Эпенетос А.А. и др.Специфическая локализация и визуализация амилоидных отложений in vivo с использованием меченного 123I амилоидного P-компонента сыворотки. J Exp Med . 1 марта 1988 г. 167 (3): 903-13. [Медлайн].
Nienhuis HL, Bijzet J, Hazenberg BP. Распространенность и лечение системного амилоидоза в западных странах. Почки (Базель) . 2016 Апрель 2 (1): 10-9. [Медлайн]. [Полный текст].
Саид С.М., Сетхи С., Валери А.М., Чанг А., Наст С.С., Краль Л. и др. Характеристика и исходы амилоидоза, связанного с хемотаксическим фактором 2 почечных лейкоцитов. Почки Инт . 2014 августа 86 (2): 370-7. [Медлайн].
Castano E, Palmer MB, Vigneault C, Luciano R, Wong S, Moeckel G. Сравнение отложения амилоида при биопсии почек человека как предиктора неблагоприятного исхода для пациента. БМК Нефрол . 2015 29 апр. 16:64. [Медлайн].
Ларсен С.П., Коссманн Р.Дж., Беггс М.Л., Соломон А., Уокер П.Д. Клинические, морфологические и генетические особенности амилоидоза хемотаксического фактора 2 лейкоцитов почек. Почки Инт . 2014 Август 86 (2): 378-82. [Медлайн].
Цыпленок ММ. Амилоидоз Alect2: primum non nocere (во-первых, не навреди). Почки Инт . 2014 Август 86 (2): 229-32. [Медлайн].
von Hutten H, Mihatsch M, Lobeck H, et al. Распространенность и происхождение амилоида при биопсии почек. Am J Surg Pathol . 2009 августа 33 (8): 1198-205. [Медлайн].
Toros AB, Erdenen F, Sari ND, Gokcay S.Почечная и надпочечная недостаточность, вторичная по отношению к семейной средиземноморской лихорадке, связанной с амилоидозом: отчет о клиническом случае. J Медицинские отчеты о случаях заболевания . 2011 18 августа. 5 (1): 390. [Медлайн]. [Полный текст].
Andronesi AG, Ismail G, Gherghiceanu M, Mitroi G, Hârza MC. Семейный амилоидоз почек, связанный со средиземноморской лихорадкой: отчет о болезни и обзор литературы. Rom J Morphol Embryol . 2019. 60 (4): 1299-1303. [Медлайн]. [Полный текст].
Benson MD, Liepnieks JJ, Yazaki M, Yamashita T, Hamidi Asl K, Guenther B, et al.Новый наследственный амилоидоз человека: результат мутации стоп-кодона в гене аполипопротеина AII. Геномика . 2001 15 марта. 72 (3): 272-7. [Медлайн].
Talamo G, Mir Muhammad A, Pandey MK, Zhu J, Creer MH, Malysz J. Оценка суточной протеинурии у пациентов с амилоидозом с использованием соотношения протеина и креатинина в случайных образцах мочи. Редкие опухоли . 2015 11 февраля. 7 (1): 5686. [Медлайн].
Purysko AS, Westphalen AC, Remer EM, Coppa CP, Leão Filho HM, Herts BR.Визуализация проявлений гематологических заболеваний с поражением почек и перинефрии. Рентгенография . 2016 июл-авг. 36 (4): 1038-54. [Медлайн]. [Полный текст].
Баррейрос А.П., Отто Г., Игни А. и др. Сонографические признаки амилоидоза. Z Гастроэнтерол . 2009 августа 47 (8): 731-9. [Медлайн].
Хокинс PN. Исследования с использованием радиоактивно меченного компонента амилоида P сыворотки крови предоставляют доказательства оборота и регрессии амилоидных отложений in vivo. Clin Sci (Колхия) . 1994 Сентябрь 87 (3): 289-95. [Медлайн].
Rydh A, Suhr O, Hietala SO, et al. Сцинтиграфия с амилоидным P-компонентом сыворотки при семейной амилоидной полинейропатии: регресс висцерального амилоида после трансплантации печени. евро J Nucl Med . 1998 25 июля (7): 709-13. [Медлайн].
Puchtler H, Sweat F, Levine M. О связывании конго красного амилоидом. Дж. Histochem Cytochem . 1962. 10: 355-64.
Sethi S, Vrana JA, Theis JD, Leung N, Sethi A, Nasr SH и др. Лазерная микродиссекция и протеомика на основе масс-спектрометрии помогают в диагностике и типировании амилоидоза почек. Почки Инт . 2012 г., 11 апреля [Medline].
Цыпленок ММ. Протеомика и масс-спектрометрия в диагностике амилоидоза почек. Клин Почки J . 2015 Декабрь 8 (6): 665-72. [Медлайн]. [Полный текст].
Sherif AM, Refaie AF, Sobh MA.Отдаленный исход трансплантации почки живого донора по поводу амилоидоза почек. Am J Kidney Dis . 2003 август 42 (2): 370-5. [Медлайн].
Gillmore JD, Booth DR, Rela M, et al. Лечебная трансплантация печени при системном амилоидозе, вызванном вариантом альфа-цепи фибриногена Glu526Val в английской семье. QJM . 2000 Май. 93 (5): 269-75. [Медлайн].
Holmgren G, Ericzon BG, Groth CG, et al. Клиническое улучшение и регресс амилоида после трансплантации печени при наследственном транстиретиновом амилоидозе. Ланцет . 1993 г., 1. 341 (8853): 1113-6. [Медлайн].
Стангу А.Дж., Баннер Н.Р., Хендри Б.М., Рела М., Портманн Б., Вендон Дж. И др. Наследственный фибриноген Амилоидоз с альфа-цепью: фенотипическая характеристика системного заболевания и роль трансплантации печени. Кровь . 15 апреля 2010 г. 115 (15): 2998-3007. [Медлайн].
Zeldenrust S, Gertz M, Uemichi T. Ортотопическая трансплантация печени при наследственном амилоидозе фибриногена. Трансплантация . 2003 27 февраля. 75 (4): 560-1. [Медлайн].
Koike H, Ando Y, Ueda M, et al. Отличительные характеристики амилоидных отложений при семейной амилоидной полинейропатии с ранним и поздним началом транстиретина Val30Met. J Neurol Sci . 24 августа 2009 г. [Medline].
Koike H, Morozumi S, Kawagashira Y, et al. Значение синдрома запястного канала в семейной амилоидной полинейропатии транстиретина Val30Met. Амилоид .2009 15 июля. 1-7. [Медлайн].
Amarzguioui M, Mucchiano G, Haggqvist B, et al. Обширные отложения амилоида, производного от аполипопротеина A1 в интиме, у пациента с мутацией аполипопротеина A1. Биохимия Биофиз Рес Коммуна . 1998, 26 января, 242 (3): 534-9. [Медлайн].
Амилоидоз — NHS
Амилоидоз — это группа редких серьезных заболеваний, вызванных накоплением аномального белка, называемого амилоидом, в органах и тканях по всему телу.
Накопление амилоидных белков (отложений) может затруднить правильную работу органов и тканей. Без лечения это может привести к органной недостаточности.
Эта страница посвящена амилоидозу AL, который является наиболее распространенным типом, и амилоидозу ATTR, который часто передается в семье.
Для получения информации о других типах амилоидоза посетите сайт информации для пациентов Национального центра амилоидоза UCL.
Симптомы амилоидоза AL
Симптомы амилоидоза AL зависят от пораженных тканей и органов.
Почечная недостаточность
У большинства людей с амилоидозом AL накапливается амилоидный белок в почках, и они подвержены риску почечной недостаточности.
Симптомы почечной недостаточности включают:
- отек, часто в ногах, вызванный задержкой жидкости (отек)
- усталость
- слабость
- потеря аппетита
Сердечная недостаточность
Отложения амилоида в сердце могут заставляют мышцы становиться жестче, что затрудняет перекачивание крови по телу.Это может привести к сердечной недостаточности, которая может вызвать:
Другие симптомы
Амилоидные белки также могут накапливаться в других областях, таких как печень, селезенка, нервы или пищеварительная система. Симптомы могут включать:
AL амилоидоз не влияет на мозг, поэтому он не вызывает, например, проблем с памятью или мышлением.
Обратитесь к терапевту, если у вас есть какие-либо из вышеперечисленных симптомов и вы беспокоитесь. Врач общей практики может назначить лечение, чтобы помочь с этими симптомами.
В некоторых случаях амилоидоз AL может быть связан с типом рака кости, называемым множественной миеломой.
Благотворительная организация Myeloma UK предоставляет дополнительную информацию о множественной миеломе и ее связи с амилоидозом AL.
Причина амилоидоза AL
Амилоидоз AL вызывается аномалией в определенных клетках костного мозга, называемых плазматическими клетками.
Аномальные плазматические клетки продуцируют аномальные формы белков легкой цепи, которые попадают в кровоток и могут образовывать амилоидные отложения.
У здоровых людей в крови нормальные белки легкой цепи, которые являются частью их естественных белков антител.Они помогают защитить организм от инфекции.
Аномальные легкие цепи у пациентов с амилоидозом AL собираются вместе в нитевидные нити (амилоидные фибриллы), от которых организм не может легко избавиться.
Со временем амилоидные фибриллы накапливаются в виде отложений амилоида AL в тканях и органах. Это постепенно останавливает их нормальное функционирование, вызывая многие симптомы амилоидоза AL.
В отличие от некоторых других типов амилоидоза, амилоидоз AL не передается по наследству, поэтому человек с этим заболеванием не может передать его своим детям.
Лечение амилоидоза AL
В настоящее время лекарства от амилоидоза не существует. Амилоидные отложения нельзя удалить напрямую.
Но есть методы лечения, которые останавливают производство аномальных белков и помогают избавиться от симптомов.
Эти процедуры могут дать вашему организму время постепенно очистить отложения, прежде чем они снова начнут накапливаться. Это может помочь предотвратить повреждение органов.
В большинстве случаев лечение будет включать химиотерапию.Химиотерапия повреждает аномальные клетки костного мозга и останавливает их производство аномальных белков, которые образуют отложения амилоида.
Стероиды обычно назначаются вместе с химиотерапией для усиления эффекта химиотерапевтических препаратов. Они также могут снизить ваши шансы на плохую реакцию на химиотерапию.
Ваш врач может также обсудить использование других методов лечения, таких как трансплантация стволовых клеток.
Вам также могут потребоваться специальные лекарства, если у вас сердечная или почечная недостаточность.
Вашим врачам и медсестрам необходимо будет тщательно контролировать количество употребляемой вами соли и количество выпиваемой вами жидкости. Вам также может потребоваться диализ, если у вас терминальная стадия почечной недостаточности.
Некоторым людям с почечной недостаточностью может подойти пересадка почки. Но основную проблему с костным мозгом все равно необходимо лечить с помощью химиотерапии, поскольку это предотвратит накопление амилоида в новой почке.
После химиотерапии вам понадобятся регулярные осмотры каждые 6–12 месяцев для выявления признаков возвращения амилоидоза AL.Если он все же вернется, возможно, вам придется снова начать химиотерапию.
Диагностика амилоидоза AL
Диагностика амилоидоза AL может быть сложной задачей, поскольку симптомы часто расплывчаты.
Небольшой образец ткани (биопсия) может быть взят из пораженной части вашего тела. Ваш врач расскажет вам, как это будет сделано.
Биопсия будет исследована под микроскопом в лаборатории, чтобы увидеть, есть ли в ней отложения амилоида.
Другие тесты
Вы также можете пройти другие тесты, чтобы оценить, как отложения амилоида повлияли на ваши отдельные органы.
Например:
- взятие образца костного мозга
- ультразвуковое сканирование сердца (эхокардиограмма) для проверки состояния вашего сердца
- различные анализы крови для выявления повреждений сердца, почек или других органов
- компьютерная томография или магнитно-резонансная томография для проверки состояния различных органов вашего тела
Национальный центр амилоидоза Национальной службы здравоохранения при Королевской бесплатной больнице в Лондоне также предлагает тип сканирования тела, называемый сканированием SAP.
Это включает введение небольшого количества радиоактивно меченного белка крови, называемого сывороточным амилоидным P-компонентом (SAP). Затем вас сканируют специальной камерой, которая обнаруживает радиоактивность.
Белок с радиоактивной меткой прикрепляется к любым отложениям амилоида в вашем теле, поэтому вы можете видеть пораженные участки тела.
Амилоидоз ATTR
АмилоидозATTR — очень редкое заболевание, вызванное отложениями амилоида из аномальных версий белка крови, называемого транстиретином (TTR).
Амилоидоз ATTR может передаваться в семье и известен как наследственный амилоидоз ATTR. Люди с наследственным амилоидозом ATTR несут мутации в гене TTR.
Это означает, что их тела на протяжении всей жизни вырабатывают аномальные белки TTR, которые могут образовывать амилоидные отложения. Обычно они поражают нервы или сердце, или и то, и другое.
Другой тип амилоидоза ATTR не является наследственным. Это называется ATTR-амилоидозом дикого типа или старческим системным амилоидозом.
В этом состоянии отложения амилоида в основном поражают сердце, а также могут вызывать синдром запястного канала у некоторых людей.
Наследственный амилоидоз ATTR может вызывать симптомы в любом возрасте от 30 лет. Симптомы ATTR-амилоидоза дикого типа обычно появляются только в возрасте примерно 65 лет.
ATTR-амилоидоз можно диагностировать с помощью:
Лечение амилоидоза ATTR
Лекарства, используемые для лечения ATTR, включают:
- патизиран (Onpattro) — данные показывают, что он снижает инвалидность и улучшает качество жизни. Он также может остановить и потенциально обратить вспять болезнь
- inotersen (Тегседи) — данные показывают, что он замедляет прогрессирование болезни
Некоторые типы наследственного амилоидоза ATTR можно лечить с помощью трансплантации печени.
Сердечную недостаточность можно лечить, тщательно контролируя количество выпиваемой и выпиваемой соли, а также принимая лекарства от сердечной недостаточности. Трансплантация сердца может быть вариантом очень редко.
Информация о вас
Если у вас амилоидоз, ваша клиническая бригада передаст информацию о вас в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния.Вы можете отказаться от регистрации в любое время.
Узнать больше о реестре
Последняя проверка страницы: 17 сентября 2020 г.
Срок следующей проверки: 17 сентября 2023 г.
NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Beers MH, Berkow R., eds. Руководство Merck, 17-е изд. Станция Уайтхаус, Нью-Джерси: Исследовательские лаборатории Мерк; 1999: 219-220.
Берков Р., изд. Руководство Merck для дома. Станция Уайтхаус, Нью-Джерси: Исследовательские лаборатории Мерк; 1997: 690-91.
Бенсон Мэриленд. Амилоидоз. В: Scriver CR, et al., Eds. Метаболические и молекулярные основы наследственных заболеваний. 7-е изд. Нью-Йорк, штат Нью-Йорк; McGraw-Hill Companies, Inc; 1995: 4159-91.
Каткарт ES. Амилоидоз. В: Kelley WN, et al., Eds. Учебник ревматологии. 4-е изд. Филадельфия, Пенсильвания: W.B. Компания Сондерс; 1993: 1413-28.
СТАТЬИ ЖУРНАЛА
Стоппини М., Беллотти В. Системный амилоидоз: уроки B2-микроглобулина. J Biol Chem. 2015; 290: 9951-9958.
Герц М.А., Бенсон М.Д., Дайк П.Дж. и др.Диагностика, прогноз и терапия транстиретинового амилоидоза. J Am Coll Cardiol. 2015; 66: 2451-2466.
Махмуд С., Палладини Дж., Санчоравала В., Вечалекар А. Последние сведения о лечении амилоидоза легких цепей. Haematologica. 2014; 99: 209-221.
Falk RH, Dubrey SW. Амилоидный порок сердца. Прогресс сердечно-сосудистых заболеваний. 2010; 52: 347-361.
Comenzo RL. Как я лечу амилоидоз. Blood 2009; 114: 3147-57
Palladini G, Merlini G. Текущее лечение амилоидоза AL.Haematologica 2009; 94: 1044-1048.
Дембер Л. Заболевание почек, связанное с амилоидозом. Варенье. Soc. Нефрол. 2006; 17: 3458-3471.
Герц MA, Lacy MQ, Dispenzieri A, Hayman SR. Амилоидоз. Передовая практика и исследования в области клинической гематологии. 2005; 18 (4): 709-727.
Бенсон М. Наследственные амилоидозы. Передовая практика и исследования в клинической ревматологии. 2003; 17: 909-927.
Park KI, Ourednik J, Ourednik V и др. Глобальные стратегии замены генов и клеток с помощью стволовых клеток.Gene Ther. 2002; 9: 613-24.
Рокен С., Шекспир А. Патология, диагностика и патогенез амилоидоза АА. Арка Вирхова. 2002; 440: 111-22.
Гринберг С.М. Церебральная амилоидная ангиопатия и дисфункция сосудов. Cerebrovasc Dis. 2002; 13 (приложение 2): 42-47.
Герц М.А., Лейси М.К., Диспензиери А. Амилоидоз легкой цепи иммуноглобулина и почки. Kidney Int. 2002; 61: 1-9.
Jaikaran ET, Clark A. Islet амилоид и диабет 2 типа; от молекулярного неправильного свертывания к патофизиологии островков.Biochim Biophys Acta. 2001; 1537: 179-203.
Санчоравала V, Райт Д.Г., Селдин округ Колумбия и др. Обзор использования высоких доз мелфалана с трансплантацией аутологичных стволовых клеток для лечения амилоидоза AL. Пересадка костного мозга. 2001; 28: 637-42.
Скиннер М., Санчоравала В., Селдин, округ Колумбия, Дембер Л.М., Фальк Р.Х., Берк Д.Л., Андерсон Дж.Дж., О’Хара С.Дж., Финн К.Т., Либби, Калифорния, Висман Дж., Квиллен К., Суон Н., Райт Д.Г.: Мелфалан в высоких дозах. и трансплантация аутологичных стволовых клеток пациентам с амилоидозом AL: 8-летнее исследование.Анналы Int Med, 2004; 140: 85-93.
Селдин Д.К., Андерсон Дж. Дж., Санчоравала В., Малек К., Райт Д. Г., Квиллен К., Финн К. Т., Берк Дж. Л., Дембер Л. М., Фальк Р. Х., Скиннер М.: Улучшение качества жизни пациентов с амилоидозом AL, получавших высокие дозы мелфалана и трансплантация аутологичных стволовых клеток. Кровь, 2004; 104: 1888-1893.
Хан MF, Falk RH. Амилоидоз. Postgrad Med J. 2001; 77: 686-93.
Добсон СМ. Сворачивание белка и его связь с болезнями человека. Biochem Soc Sympos. 2001; 68: 1-26.
Адамс Д. Наследственные и приобретенные амилоидные невропатии. J Neurol. 2001; 248: 647-57.
Сараива MJ. Транстиретиновый амилоидоз: рассказ о слабых взаимодействиях. FEBS Lett. 2001; 498: 201-203.
Floege J, Schaffer J, Koch KM. Сцинтиграфические методы выявления бета2-микроглобулинового амилоидоза (амилоидоз Abeta2-микроглобулина). Пересадка нефрола Dial. 2001; 16 (приложение 4): 12-16.
Walker LC, LeVine H. Церебральные протеопатии: нейродегенеративные нарушения конформации и сборки белков.Mol Neurobiol. 2000; 21: 83-95.
Эль-Шанти HE. Семейная средиземноморская лихорадка. Саудовская медицина, 2001; 22: 104-9.
Cunnane G. Предшественники амилоида и амилоидоз при воспалительном артрите. Curr Opin Rheumatol. 2001; 13: 67-73.
Plante-Bordeneuve V, Said G. Транстиретиновая родственная знакомая амилоидная полинейропатия. Curr Opin Neurol. 2000; 13: 569-73.
Герц М.А., Коменцо Р., Фальк Р.Х. и др. Определение вовлечения органов и ответа на лечение при амилоидозе легкой цепи иммуноглобулина (AL): согласованное мнение 10-го Международного симпозиума по амилоиду и амилоидозу.Являюсь. J. Hematol. 2005; 79: 319-328.
ИНТЕРНЕТ
ИНФОРМАЦИЯ О амилоидозе: для пациентов и их сети поддержки, включая врачей, медсестер и студентов-медиков. Группы поддержки амилоидоза. Октябрь 2013 г. http://www.amyloidosissupport.com/AmyloidAware_Booklet.pdf Проверено 24 октября 2018 г.
Basu A, Bogdan CA, Matute R. Бета-2m амилоидоз, связанный с диализом. Medscape. Обновлено: 3 февраля 2017 г. Доступно по адресу: http://emedicine.medscape.com/article/246542-overview Проверено 24 октября 2018 г.
Что такое амилоидоз? Программа лечения и исследования амилоидов Бостонского университета. http://www.bu.edu/amyloid/about/what/ По состоянию на 24 октября 2018 г.
Amyloidosis. Фонд Мэйо медицинского образования и исследований. 7 июля 2017 г. http://www.mayoclinic.com/health/amyloidosis/DS00431 По состоянию на 24 октября 2018 г.
Амилоидная нефропатия | Клинический журнал почек
Аннотация
Амилоидоз — необычное заболевание, которое характеризуется аномальным внеклеточным отложением неправильно свернутых белковых фибрилл, приводящим к дисфункции органов.Депонированные белки обладают общими химическими и гистологическими свойствами, но могут сильно различаться по своему происхождению. Заболевание почек — частое проявление у пациентов с системным амилоидозом, при этом в образцах биопсии почек обнаружен ряд амилоидогенных белков. Появление протеомики на основе масс-спектрометрии повысило диагностическую точность и общее понимание амилоидоза. В этом подробном обзоре обсуждаются общие гистопатологические особенности почечного амилоидоза и подробно обсуждаются конкретные формы амилоида, поражающего почки.
Введение
Амилоидоз представляет собой семейство заболеваний, определяемых внеклеточным отложением белковых фибрилл с характерной конформацией β-складчатых пластин. Заболевание почек — частое проявление и основная причина заболеваемости у этих пациентов. Рудольф Вирхов впервые использовал термин амилоид в 1854 году [1], а в 1971 году моноклональная легкая цепь стала первым химически охарактеризованным амилоидным белком [2]. Протеомика на основе масс-спектрометрии значительно расширила возможности диагностики и определения амилоида, и на сегодняшний день идентифицировано до 30 амилоидогенных белков [1,3,4].Хотя многие белки вовлечены в амилоидоз, все амилоидные фибриллы обладают общими химическими и ультраструктурными свойствами, включая организацию в нерастворимые бета-складчатые листы, идентифицированные с помощью рентгеновской кристаллографии [5, 6]. Этот обзор будет сосредоточен на почечных проявлениях амилоидоза, включая патофизиологические механизмы, гистологические особенности, идентификацию белков, а также морфологические и клинические характеристики общих и редких форм амилоидоза.
Гистологическая диагностика и идентификация амилоида
Гистологические особенности амилоида одинаковы независимо от его состава.На срезах ткани, окрашенных H и E, амилоид идентифицируется как внеклеточный аморфный материал, который является слегка эозинофильным. Эти отложения часто слабо окрашивают периодическую кислоту Шиффа (PAS), демонстрируют оттенок от синего до серого на окрашивании трихромом и обычно отрицательны при окрашивании метенамином серебра Джонса (JMS) (рис. 1). Эти тинкториальные свойства контрастируют с гистологическим внешним видом коллагена, основного компонента базальных мембран, мезангиального матрикса и участков склероза, что демонстрирует сильную положительную реакцию на ПАВ и ЮМС.Все типы амилоида проявляют сродство к красителю Конго красный, демонстрируя оранжево-красный вид при световой микроскопии и характерное яблочно-зеленое двойное лучепреломление при поляризации (рис. 2). Для получения оптимальных результатов срезы тканей для окрашивания конго красным необходимо нарезать толще (> 5 мкм), чем срезы размером 2–4 мкм, используемые для обычных красителей с помощью световой микроскопии. Тиофлавин Т также связывает структуры, богатые бета-слоями, но менее специфичен, чем конго красный [3]. По данным электронной микроскопии, амилоидные отложения состоят из случайно ориентированных неразветвленных фибрилл диаметром от 7 до 10 нм.
Рис. 1.
Амилоид обычно имеет слегка эозинофильный вид при окрашивании гематоксилином и эозином ( A ), слабом окрашивании PAS ( B ), отрицательном окрашивании серебром ( C ) и демонстрирует сине-серый цвет. оттенок на морилке трихром ( D ).
Рис. 1.
Амилоид обычно проявляет слегка эозинофильный вид при окрашивании гематоксилином и эозином ( A ), слабом окрашивании PAS ( B ), отрицательном окрашивании серебром ( C ) и демонстрирует синий цвет. серый оттенок на морилке трихром ( D ).
Рис. 2.
Исследование Конго красным ( A ) подчеркивает глобальное мезангиальное и сегментарное окрашивание стенок капилляров аморфными отложениями. После поляризации ( B ) конгофильный материал демонстрирует характерное двойное лучепреломление яблочно-зеленого цвета, что подтверждает присутствие амилоида.
Рис. 2.
Исследование Конго красным ( A ) подчеркивает глобальное мезангиальное и сегментарное окрашивание стенок капилляров аморфными отложениями. После поляризации ( B ) конгофильный материал демонстрирует характерное двойное лучепреломление яблочно-зеленого цвета, что подтверждает присутствие амилоида.
В почках отложения амилоида могут быть обнаружены в любом из паренхиматозных отделов, включая клубочки, канальцы, интерстиций и / или сосуды. Чаще всего поражаются клубочки, 97% случаев в исследовании показывают отложение клубочков [7]. В большинстве случаев накопление амилоида вовлекает мезангиум перед стенками капилляров. В ранних случаях процесс может быть незаметным и затрагивать только несколько мезангиальных областей, поэтому его легко пропустить при рутинной гистологической оценке.Более обширное поражение приводит к заметному расширению мезангиума, который может принимать узелковый вид и имитировать мезангиальные склеротические процессы, такие как диабетический гломерулосклероз. Однако часто отрицательное окрашивание PAS и JMS более характерно для амилоида. Иногда субэпителиальные или внутримембранные отложения амилоида, состоящие из фибрилл, расположенных перпендикулярно базальной мембране клубочков, могут обнаруживать широко расставленные субэпителиальные шипы (так называемые амилоидные спикулы) (Рисунок 3).Эти спикулы содержат серебро и имеют тенденцию быть более локальными и более длинными, чем «шипы», наблюдаемые при перепончатой нефропатии. Прогрессирование отложения амилоида в конечном итоге вызывает полное стирание клубочковой архитектуры. Поражение клубочков часто сопровождается отложениями в тубулоинтерстиции и / или почечных сосудах, особенно в межлобулярных артериях и артериолах. Локализация амилоида влияет на клиническую картину. В случаях с преимущественным поражением клубочков протеинурия часто преобладает, тогда как почечная недостаточность может быть основным проявлением, когда тубулоинтерстициальные или сосудистые отложения более многочисленны.В редких случаях почечные мозговые отложения являются значительными и приводят к дефектам концентрации мочи с полиурией и никтурией. Как это часто бывает, отложения амилоида можно увидеть в нескольких отделах со смешанной клинической картиной.
Рис. 3.
Фокальные субэпителиальные спикулы видны на большом увеличении ( A , JMS). Ультраструктурное исследование ( B ) обнаруживает обильные амилоидные фибриллы, которые плотно упакованы в спикулах (маленькое изображение).
Рис. 3.
Фокальные субэпителиальные спикулы видны на большом увеличении ( A , JMS). Ультраструктурное исследование ( B ) обнаруживает обильные амилоидные фибриллы, которые плотно упакованы в спикулах (маленькое изображение).
Хотя все формы амилоидоза имеют общие гистологические и химические свойства, патогенез и стратегия лечения могут значительно различаться в зависимости от белка-предшественника. Типирование часто можно выполнить с помощью рутинной иммунофлуоресценции или иммуногистохимического окрашивания при более распространенных формах амилоидоза.Иммунофлуоресцентное окрашивание на иммуноглобулины (Ig) и легкие цепи обычно выполняется на образцах биопсии почек и в большинстве случаев позволяет идентифицировать монотипические легкие цепи (рис. 4). В отдельных случаях амилоидоз легких цепей (AL) может показывать неубедительное или отрицательное окрашивание [7–9]. Обычные иммунофлуоресцентные антитела для легких цепей каппа и лямбда могут нацеливаться на домены, которые могут быть изменены или удалены в амилоидогенных формах. Исследования показали, что до 36% AL в почках не окрашиваются легкими цепями [10].
Рис. 4.
Иммунофлуоресцентная микроскопия в случае амилоидоза легкой цепи показывает сильное окрашивание аморфных гломерулярных отложений на легкую лямбда-цепь ( A ) без окрашивания на легкую каппа-цепь ( B ).
Рис. 4.
Иммунофлуоресцентная микроскопия в случае амилоидоза легкой цепи показывает сильное окрашивание аморфных гломерулярных отложений на легкую лямбда-цепь ( A ) без окрашивания на легкую каппа-цепь ( B ).
Недавнее появление лазерной микродиссекции и протеомного анализа на основе масс-спектрометрии (LMD / MS) оказалось точным и полезным инструментом для типирования амилоида [4]. Said et al. сообщил о самой крупной на сегодняшний день серии амилоидоза почек, а происхождение амилоида было обнаружено в> 97% случаев с помощью LMD / MS. Идентификация амилоида на основе протеомики рекомендуется в тех случаях, когда рутинное иммунофлуоресцентное и / или иммуногистохимическое тестирование не может однозначно определить амилоидные отложения (Таблица 1) [4, 7].LMD / MS требует помощи опытной лаборатории, а подробные методы LMD / MS были ранее опубликованы [4, 11]. Вкратце, LMD / MS выполняется, сначала идентифицируя конго-положительные области в биопсии, которые могут включать клубочки, стенки сосудов и / или интерстициальные отложения. Эти очаги подвергаются микродиссекции и удаляются из ядра ткани с последующим расщеплением до триптических пептидов. Полученные пептиды анализируют с использованием тандемной масс-спектрометрии с электрораспылением жидкостной хроматографии и генерируют список белков из пептидных последовательностей.Общее количество собранных масс-спектров, сопоставленных с конкретным белком, указывается значением «спектров» (рис. 5). Более высокие значения спектров указывают на большее количество белка и большую уверенность в точности идентификации белка [4, 11].
Таблица 1.Рекомендуемые показания для использования протеомики на основе лазерной микродиссекции / масс-спектрометрии для амилоидного типирования в биопсии почек
| Отсутствие ткани, доступной для иммунофлуоресцентной микроскопии |
| Отрицательное иммунофлуоресцентное окрашивание и отрицательное иммунофлуоресцентное окрашивание для каппа-цепей иммуногистохимическое окрашивание сывороточного амилоидного белка A |
| Равное иммунофлуоресцентное окрашивание для легких цепей каппа и лямбда. |
| Сильное иммунофлуоресцентное окрашивание тяжелых цепей иммуноглобулина с окрашиванием легких цепей или без него. |
| Положительное иммунофлуоресцентное окрашивание на легкие цепи каппа и / или лямбда И положительное иммуногистохимическое окрашивание на сывороточный амилоидный белок. |
| Сомнительное окрашивание конго красным. |
| Отсутствие ткани, доступной для иммунофлуоресцентной микроскопии | ||
| Отрицательное иммунофлуоресцентное окрашивание для каппа- и лямбда-легких цепей И отрицательное иммуногистохимическое окрашивание для сывороточного амилоидного A-протеина | a иммунофлуоресценция | a 912 цепи. |
| Сильное иммунофлуоресцентное окрашивание тяжелых цепей иммуноглобулина с окрашиванием легких цепей или без него. | ||
| Положительное иммунофлуоресцентное окрашивание на легкие цепи каппа и / или лямбда И положительное иммуногистохимическое окрашивание на сывороточный амилоидный белок. | ||
| Сомнительное окрашивание конго красным. |
Рекомендуемые показания к применению протеомики на основе лазерной микродиссекции / масс-спектрометрии для амилоидного типирования при биопсии почек
| Отсутствие ткани, доступной для иммунофлуоресцентной микроскопии |
| a легкие цепи И отрицательное иммуногистохимическое окрашивание сывороточного амилоида A белка |
| Равное иммунофлуоресцентное окрашивание для легких цепей каппа и лямбда. |
| Сильное иммунофлуоресцентное окрашивание тяжелых цепей иммуноглобулина с окрашиванием легких цепей или без него. |
| Положительное иммунофлуоресцентное окрашивание на легкие цепи каппа и / или лямбда И положительное иммуногистохимическое окрашивание на сывороточный амилоидный белок. |
| Сомнительное окрашивание конго красным. |
| Отсутствие ткани, доступной для иммунофлуоресцентной микроскопии | ||
| Отрицательное иммунофлуоресцентное окрашивание для каппа- и лямбда-легких цепей И отрицательное иммуногистохимическое окрашивание для сывороточного амилоидного A-протеина | a иммунофлуоресценция | a 912 цепи. |
| Сильное иммунофлуоресцентное окрашивание тяжелых цепей иммуноглобулина с окрашиванием легких цепей или без него. | ||
| Положительное иммунофлуоресцентное окрашивание на легкие цепи каппа и / или лямбда И положительное иммуногистохимическое окрашивание на сывороточный амилоидный белок. | ||
| Сомнительное окрашивание конго красным. |
Рис. 5.
Типичные данные масс-спектроскопии в случае амилоидоза LECT2.Анализ показывает высокие спектры LECT2, аполипопротеина E и сывороточного амилоидного P-компонента в амилоидных отложениях. Предоставлено доктором Сандживом Сетхи (клиника Мэйо, Рочестер, Миннесота, США).
Рис. 5.
Типичные данные масс-спектроскопии в случае амилоидоза LECT2. Анализ показывает высокие спектры LECT2, аполипопротеина E и сывороточного амилоидного P-компонента в амилоидных отложениях. Предоставлено доктором Сандживом Сетхи (клиника Мэйо, Рочестер, Миннесота, США).
Амилоидозы почек
Многие амилоидогенные белки могут вызывать заболевание почек (таблица 2).Механизмы амилоидогенеза в этих случаях вариабельны и включают аномальную продукцию белка, перепроизводство или снижение экскреции белков дикого типа и наследственные мутации. Наиболее распространенный тип амилоидоза почек связан с дискразией плазматических клеток и состоит из компонентов Ig. Чаще всего они происходят из моноклональной легкой цепи (AL), но редко состоят из тяжелой цепи Ig (AH) или как тяжелой, так и легкой цепи Ig (AHL) [13, 14]. Амилоидоз, возникающий из-за сывороточного амилоида A (SAA), представляет собой второй по распространенности тип амилоида (после AL) и обычно возникает при хронических воспалительных состояниях.О амилоидогенном хемотаксическом факторе 2 лейкоцитов (ALECT2) сообщалось в двух больших сериях как о третьей наиболее распространенной форме почечного амилоида; однако это открытие, вероятно, зависит от географического положения, учитывая его более высокую распространенность у пациентов мексиканского происхождения [7, 15]. Другие формы почечного амилоида включают цепь Aα фибриногена (AFib), лизоцим (ALys), аполипопротеины AI (AApo AI), AII (AApo AII), AIV (AApo AIV), транстиретин (ATTR) и гельсолин (AGel).
Таблица 2.Зарегистрированные белки-предшественники при амилоидозе почек
| Тип амилоида . | Распределение почек . | Экстрапочечное поражение . |
|---|---|---|
| Ig-связанные (AL, AH и AHL) | Все компартменты: наиболее часто встречающиеся клубочки | Все органы, за исключением центральной нервной системы |
| AA | Все компартменты: наиболее часто встречающиеся клубочки | 912 Все органы, за исключением центральной нервной системы|
| ALect2 | Все компартменты: наиболее часто встречающийся интерстиций | Легкое, печень, надпочечники, селезенка и толстая кишка |
| AFib | Отложения в клубочках: внегломерулярные отложения встречаются реже | Надпочечники, селезенка и периферическая нервная система |
| ALys | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Печень, желудочно-кишечный тракт, селезенка, лимфатический узел, кожа и слюнная железа |
| Печень, сердце, кожа, гортань, небо, периферическая нервная система и семенники | ||
| AApo AII | Все отделы: клубочки и мелкие сосуды, наиболее часто встречающиеся | Надпочечники и мелкие сосуды других органов |
| AApo AIV | Медуллярный интерстиций | Сердце |
| ATTR | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Периферическая нервная система, вегетативная нервная система, сердце, желудочно-кишечный тракт и глаз |
| AGel | Преимущественно клубочковые | Роговица и кожа|
| Амилоидный тип . | Распределение почек . | Экстрапочечное поражение . |
|---|---|---|
| Ig-связанные (AL, AH и AHL) | Все компартменты: наиболее часто встречающиеся клубочки | Все органы, за исключением центральной нервной системы |
| AA | Все компартменты: наиболее часто встречающиеся клубочки | 912 Все органы, за исключением центральной нервной системы|
| ALect2 | Все компартменты: наиболее часто встречающийся интерстиций | Легкое, печень, надпочечники, селезенка и толстая кишка |
| AFib | Отложения в клубочках: внегломерулярные отложения встречаются реже | Надпочечники, селезенка и периферическая нервная система |
| ALys | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Печень, желудочно-кишечный тракт, селезенка, лимфатический узел, кожа и слюнная железа |
| Печень, сердце, кожа, гортань, небо, Периферическая нервная система и семенники | ||
| AApo AII | Все отделы: клубочки и мелкие сосуды, наиболее часто встречающиеся | Надпочечники и мелкие сосуды других органов |
| AApo AIV | Медуллярный интерстиций | a1213 Сердце|
| ATTR | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Периферическая нервная система, вегетативная нервная система, сердце, желудочно-кишечный тракт и глаз |
| AGel | Преимущественно клубочковые | Роговица и кожа|
Зарегистрированные белки-предшественники при амилоидозе почек
| Амилоидный тип . | Распределение почек . | Экстрапочечное поражение . |
|---|---|---|
| Ig-связанные (AL, AH и AHL) | Все компартменты: наиболее часто встречающиеся клубочки | Все органы, за исключением центральной нервной системы |
| AA | Все компартменты: наиболее часто встречающиеся клубочки | 912 Все органы, за исключением центральной нервной системы|
| ALect2 | Все компартменты: наиболее часто встречающийся интерстиций | Легкое, печень, надпочечники, селезенка и толстая кишка |
| AFib | Отложения в клубочках: внегломерулярные отложения встречаются реже | Надпочечники, селезенка и периферическая нервная система |
| ALys | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Печень, желудочно-кишечный тракт, селезенка, лимфатический узел, кожа и слюнная железа |
| Печень, сердце, кожа, гортань, небо, периферическая нервная система и семенники | ||
| AApo AII | Все отделы: клубочки и мелкие сосуды, наиболее часто встречающиеся | Надпочечники и мелкие сосуды других органов |
| AApo AIV | Медуллярный интерстиций | Сердце |
| ATTR | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Периферическая нервная система, вегетативная нервная система, сердце, желудочно-кишечный тракт и глаз |
| AGel | Преимущественно клубочковые | Роговица и кожа|
| Амилоидный тип . | Распределение почек . | Экстрапочечное поражение . |
|---|---|---|
| Ig-связанные (AL, AH и AHL) | Все компартменты: наиболее часто встречающиеся клубочки | Все органы, за исключением центральной нервной системы |
| AA | Все компартменты: наиболее часто встречающиеся клубочки | 912 Все органы, за исключением центральной нервной системы|
| ALect2 | Все компартменты: наиболее часто встречающийся интерстиций | Легкое, печень, надпочечники, селезенка и толстая кишка |
| AFib | Отложения в клубочках: внегломерулярные отложения встречаются реже | Надпочечники, селезенка и периферическая нервная система |
| ALys | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Печень, желудочно-кишечный тракт, селезенка, лимфатический узел, кожа и слюнная железа |
| Печень, сердце, кожа, гортань, небо, Периферическая нервная система и семенники | ||
| AApo AII | Все отделы: клубочки и мелкие сосуды, наиболее часто встречающиеся | Надпочечники и мелкие сосуды других органов |
| AApo AIV | Медуллярный интерстиций | a1213 Сердце |
| ATTR | Все отделы: клубочки и артериолы, наиболее часто встречающиеся | Периферическая нервная система, вегетативная нервная система, сердце, желудочно-кишечный тракт и глаз |
| AGel | Преимущественно клубочковые | Роговица и кожа|
AL, AH и AHL
AL — наиболее распространенная форма амилоида в развитых странах.В двух недавних исследованиях, проведенных в США и Италии, на АЛ приходится 81 и 68% случаев амилоидоза почек соответственно [7, 16]. АГ и АГЛ являются необычными формами амилоидоза и относятся к категории Ig-связанного амилоидоза. Ig-связанный амилоидоз связан с дискразией плазматических клеток и редко встречается с другими лимфопролиферативными заболеваниями. В исследовании 190 пациентов с множественной миеломой, которым проводилась биопсия почки, у 40 пациентов (21%) был выявлен амилоидоз, из которых 88% были AL [17].Легкая цепь лямбда чаще является амилоидогенным белком, чем легкая цепь каппа в соотношении 3: 1 [18, 19].
AL амилоидные фибриллы состоят либо из полностью интактных легких цепей (включая как константные, так и вариабельные домены), либо только из вариабельного домена. Мутации в N-конце (содержащемся в вариабельном домене) и подгруппе V λ VI могут приводить к усилению амилоидогенных свойств [13, 20]. Посттрансляционная модификация белка и протеолиз также могут играть роль в отложении амилоида [21].Патогенез АГ и АГЛ менее изучен, но предполагаются аналогичные механизмы [14].
Заболевание почек является наиболее частым проявлением системного Ig-связанного амилоидоза, встречающегося почти у 50% пациентов. Средний возраст постановки диагноза составляет 65 лет, и многие исследования продемонстрировали небольшое преобладание мужчин с соотношением мужчин и женщин ~ 2: 1 [7, 14, 16, 22]. Когда поражена почка, почечная недостаточность является обычным явлением, составляя 47% случаев в исследовании [7].Протеинурия зарегистрирована более чем у 73% пациентов с Ig-ассоциированным почечным амилоидом с полным нефротическим синдромом у 25–68% [13, 16]. Пациенты с AL-λ, как правило, имеют более низкий уровень креатинина в сыворотке и более высокую степень протеинурии, чем пациенты с AL-κ. По сравнению с амилоидом AL, AH и AHL чаще связаны с гематурией, но достоверных различий в уровне креатинина сыворотки, расчетной скорости клубочковой фильтрации (eGFR), содержания белка в суточной моче или процента пациентов с нефротическим синдромом не установлено.Вне почек АГ и АГЛ связаны с менее частым поражением сердца, и пациенты с большей вероятностью имеют явную множественную миелому, чем пациенты с AL [14].
Гистологически AL / AH / AHL может поражать все паренхимные компартменты почек, причем наиболее часто поражаются клубочки. Nasr et al. не обнаружил существенных различий в паренхиматозном распределении между AL и AH / AHL. Были выявлены некоторые гистологические различия, включая четыре случая (25%) AH / AHL с PAS-положительными амилоидными отложениями [14].В редких случаях отложения AL могут быть ограничены сосудистой сетью, и это открытие было связано с более высоким уровнем креатинина в сыворотке крови при предъявлении и более низкой протеинурией, чем при диффузной AL [23]. Сообщалось также о необычном случае внутриканальцевого амилоидоза AL-λ с острым повреждением почек без поражения клубочков, интерстициальной ткани или сосудов [24]. Иногда амилоидоз можно увидеть в сочетании с другими моноклональными Ig-ассоциированными заболеваниями, такими как нефропатия отложения легких цепей и болезнь отложения легких цепей [25].
Лечение Ig-связанного амилоидоза обычно направлено на лежащую в основе дискразию плазматических клеток. У нелеченных пациентов с ОЛ медиана выживаемости составляет 12 месяцев после постановки диагноза и <5 месяцев у пациентов с поражением сердца. Прогноз улучшился с развитием антиплазменной терапии и трансплантации стволовых клеток. Скиннер и др. продемонстрировал увеличенную среднюю выживаемость 4,6 года у 312 пациентов, получавших высокие дозы мелфалана с последующей трансплантацией аутологичных стволовых клеток (ASCT) [26].В исследовании Leung et al. , почечный ответ у пациентов с AL после приема высоких доз мелфалана и ASCT наблюдался у 43% пациентов и был связан со степенью исходной протеинурии [27]. Пациентам с ОЛ трансплантация почки выполнялась редко. Пинни и др. сообщил о серии, включающей 25 пациентов с AL, перенесших трансплантацию почки. Средняя выживаемость трансплантата составила 5,8 года при 5- и 10-летней выживаемости трансплантата 74 и 25% соответственно.В 28% трансплантатов обнаружен рецидив почечного амилоида; однако потеря трансплантата не была связана с амилоидной нефропатией. Во время трансплантации те, у кого не было хотя бы частичного клонального ответа на лечение, имели значительно худшую выживаемость трансплантата [28].
AA
Амилоид А амилоидоз — вторая по распространенности форма системного амилоидоза. В двух исследованиях из США АК составляла 7 и 12,5% биопсий почек с амилоидозом [4, 7]. АК более распространена в Европе, где она регистрируется у 30–40% пациентов с отложениями амилоида в почках [16, 29, 30].Амилоидные фибриллы АК происходят из белка SAA, который является аполипопротеиновым компонентом липопротеинов высокой плотности (ЛПВП) и реагентом острой фазы [31]. SAA продуцируется гепатоцитами и находится под транскрипционной регуляцией провоспалительных цитокинов [32]. У меньшинства пациентов с устойчивым воспалительным стимулом и гиперпродукцией SAA может происходить неправильная укладка белка с последующим отложением амилоида. Основные воспалительные стимулы, связанные с АК, включают хронический воспалительный артрит, инфекцию, синдромы периодической лихорадки (например, семейную средиземноморскую лихорадку), воспалительное заболевание кишечника, неоплазию и болезнь Кастлемана [29, 33–38].Lachmann et al. сообщил о среднем интервале 17 лет между началом воспалительного заболевания и диагнозом амилоидоза АА [33].
Семейство генов SAA расположено на коротком плече хромосомы 11 с тремя экспрессируемыми генными продуктами. Гены SAA1, и SAA2 преимущественно экспрессируются как часть острофазового ответа и являются первичными изотипами, депонированными в AA. Экспрессия SAA4 является конститутивно активной и не является реагентом острой фазы [32].Сообщалось о случае АА, вторичного по отношению к мутации SAA4 в отсутствие хронического воспаления [39].
Средний возраст пациентов с АА составляет ~ 10 лет, они моложе пациентов с ОЛ, и АА представляет собой наиболее частую форму амилоидоза у детей [40]. Заболевание почек является наиболее частым клиническим проявлением АК: до 97% пациентов в исследовании имели протеинурию> 500 мг в день или креатинин сыворотки> 132,6 мкмоль / л (1,5 мг / дл) [33]. Полный нефротический синдром отмечен у 54–63% пациентов, а повышенный креатинин сыворотки — у 75–81% [7, 29].
Гистологическая локализация отложений АК в паренхиме почек сходна с АЛ. Гломерулярные отложения являются наиболее распространенными, а внегломерулярные отложения, включая базальную мембрану канальцев, интерстициальные и сосудистые отложения, наблюдаются в 12, 73 и 88% случаев, соответственно, в исследовании [7]. Verine et al. сообщил о воспалительной реакции, связанной с амилоидом, в 30 из 68 случаев. Девятнадцать показали гранулематозную реакцию вокруг сосудистых и интерстициальных отложений, а 17 продемонстрировали гломерулярные эндокапиллярные лейкоциты.Двенадцать из них продемонстрировали серповидное и / или некротическое повреждение клубочка [29]. Возможность наложенного гломерулонефрита была повышена, а серологический анализ на цитоплазматические антитела против нейтрофилов был отрицательным у 8 из 12 протестированных пациентов. Гистологическую идентификацию АК можно провести с помощью иммуногистохимии (рис. 6). Иногда различие между АА и Ig-связанным амилоидозом может быть затруднено из-за различного неспецифического иммунофлюоресцентного окрашивания для компонентов Ig и комплемента в отложениях амилоида [7, 8].В таких случаях LMD / MS может помочь в идентификации белка-предшественника.
Рис. 6.
Срез, окрашенный гематоксилином и эозином ( A ), показывает накопление аморфного и слегка эозинофильного материала в клубочках, артериолах и интерстициальных областях. Окрашивание конго красным (не показано) было положительным для амилоида, а иммунофлуоресцентное окрашивание для легких цепей (не показано) было отрицательным. Иммуногистохимическое окрашивание белка SAA ( B ) подтвердило наличие амилоидоза AA.
Рис. 6.
Срез, окрашенный гематоксилином и эозином ( A ), показывает накопление аморфного и слегка эозинофильного материала в клубочках, артериолах и интерстициальных областях. Окрашивание конго красным (не показано) было положительным для амилоида, а иммунофлуоресцентное окрашивание для легких цепей (не показано) было отрицательным. Иммуногистохимическое окрашивание белка SAA ( B ) подтвердило наличие амилоидоза AA.
Лечение амилоидоза AA в первую очередь направлено на основное хроническое воспалительное состояние с целью снижения выработки SAA.Lachmann et al. показал, что отложения амилоида АК обладают потенциалом к регрессу, когда циркулирующие SAA могут поддерживаться на низком уровне [33]. Улучшение почечной функции, как правило, было медленным процессом, который часто длился от нескольких месяцев до нескольких лет; однако повторение воспалительного стимула приводило к быстрому прогрессированию заболевания почек [31, 33]. У пациентов с амилоидозом AA, вторичным по отношению к ревматическому заболеванию, иммуносупрессивная терапия была основой лечения, включая таргетную терапию против фактора некроза опухоли (TNF) и, в последнее время, интерлейкина-6 (IL-6).Okuda et al. сравнил лечение тоцилизумабом (ингибитор IL-6) и терапию анти-TNF у пациентов с амилоидозом AA, вторичным по отношению к ревматическому заболеванию. Те, кто лечился тоцилизумабом, показали больший контроль SAA, улучшение СКФ и более высокие показатели клинической ремиссии, чем те, кто получал терапию анти-TNF [41]. У пациентов с другими воспалительными состояниями лечение, направленное на конкретное заболевание, принесло пользу. К ним относятся колхицин у пациентов с семейной средиземноморской лихорадкой, антимикробная терапия у пациентов с хронической инфекцией и хирургическая резекция опухолей [34, 35].Другой многообещающий подход к лечению нацелен на само отложение амилоида. Эпродизат, который имеет структурное сходство с сульфатом гепарина, нарушает взаимодействие между амилоидогенными фибриллами и гликозаминогликанами. В многоцентровом исследовании было показано, что он замедляет прогрессирование заболевания почек при амилоидозе АА [42]. У тех, кто прогрессирует до терминальной стадии почечной недостаточности (ТПН), трансплантация почки является общепринятой формой заместительной почечной терапии. В исследовании с участием 43 пациентов с амилоидозом AA, получавших трансплантацию почки, Pinney et al. сообщил о 5- и 10-летней выживаемости трансплантата 86 и 59% соответственно. У девяти пациентов развился рецидив амилоида в трансплантате, а у двоих в результате этого трансплантата не удалось. Как и следовало ожидать, сывороточные концентрации SAA были значительно выше у пациентов с рецидивирующим амилоидом, чем у пациентов без него [28].
ALECT2
Лейкоцитарный хемотаксический фактор 2 (LECT2) является недавним дополнением к постоянно растущему списку амилоидогенных белков [43]. LECT2 продуцируется гепатоцитами и в первую очередь действует как хемотаксический фактор нейтрофилов, а также может играть роль в восстановлении и росте тканей после повреждения [44, 45].В двух недавних исследованиях, проведенных в США, ALECT2 был третьей по частоте формой амилоидоза почек, составляющей 3% случаев амилоидоза почек [7, 15]. Механизм амилоидогенеза до конца не изучен, но предполагалась его возможная генетическая роль. Мерфи и др. выполнил секвенирование генов у четырех пациентов с ALECT2 и обнаружил, что все четверо были гомозиготными по аллелю G, полученному в результате аминокислотной замены (Ile40Val) [46]. Большинство зарегистрированных случаев было выявлено у пациентов мексиканского происхождения, что также свидетельствует о генетической роли [47].Клиническое заболевание, связанное с ALECT2, в основном ограничивалось почками, причем в одном отчете демонстрируются случайно обнаруженные отложения ALECT2 в ткани печени, селезенки, толстой кишки и надпочечников [48]. Отложения ALECT2 в легких и почках проявляются как легочно-почечный синдром [49].
Почечный ALECT2 чаще проявляется почечной недостаточностью, чем тяжелой протеинурией. Said et al. сообщил о 13 случаях ALECT2 и обнаружил, что 17% имели нефротический синдром, тогда как 92% имели почечную недостаточность [7].Эта клиническая картина коррелирует с гистологическим распределением амилоидных отложений. Отложение ALECT2 наблюдается во всех почечных компартментах; однако поровые отложения часто преобладают (Рисунок 7) [7, 15, 46, 50]. Идентификация ALECT2 лучше всего выполняется LMD / MS; однако, по нашему опыту, иммуногистохимическая характеристика является полезным инструментом, если она выполняется опытной лабораторией [49].
Рис. 7.
Исследование конго красным ( A ) демонстрирует сильное окрашивание амилоидных отложений в интерстиции, сосудистой сети и мезангиальных областях.Иммуногистохимическое окрашивание ( B ) на LECT2 положительно в этих отложениях.
Рис. 7.
Исследование конго красного ( A ) демонстрирует сильное окрашивание амилоидных отложений в интерстиции, сосудистой сети и мезангиальных областях. Иммуногистохимическое окрашивание ( B ) на LECT2 положительно в этих отложениях.
Наследственный амилоидоз
Наследственные амилоидозы — это редкая, но клинически значимая и разнообразная группа заболеваний, которые в основном наследуются по аутосомно-доминантному типу.Связанные белки-предшественники включают фибриногеновую цепь Aα (AFib), лизоцим (ALys), аполипопротеины AI (AApo AI) и AII (AApo AII), транстиретин (ATTR) и гельсолин (AGel). В исследовании 350 пациентов с первоначально диагностированным AL генетический анализ показал наследственный амилоидоз в 34 случаях (9,7%) [9]. Идентификация белков-предшественников при наследственных формах амилоидоза требует высокого индекса подозрительности, учитывая, что специфические иммуногистохимические окрашивания часто недоступны. Как и другие типы амилоида, LMD / MS оказался полезным для идентификации белка-предшественника, особенно в сочетании с генетическим тестированием на конкретные мутации.
Амилоидоз цепи фибриногена Aα был впервые описан в 1993 г. и в первую очередь проявляется как прогрессирующее заболевание почек [51]. По крайней мере, девять мутаций были идентифицированы с вариантом Glu526Val у 90% пациентов в исследовании [52]. Амилоидные фибриллы состоят из фрагментов карбоксильного конца цепи Aα фибриногена со всеми известными мутациями, затрагивающими эту область [52, 53]. Gillmore et al. сообщил о самой большой на сегодняшний день серии пациентов со средним возрастом на момент обращения 58 лет.Протеинурия была универсальным проявлением, почечная недостаточность присутствовала в 54% случаев [52]. Биопсия почки часто показывает заметное отложение клубочков с частой облитерацией просвета капилляра и незначительным внегломерулярным отложением или его отсутствием [7, 52, 54, 55]. Иммунофлуоресцентная микроскопия может быть полезна для диагностики AFib с узловым мезангиальным окрашиванием на фибриноген, что позволяет предположить диагноз [54]. Внепочечные отложения встречаются редко и в основном возникают на поздних стадиях течения болезни. Большинство пациентов следуют прогрессирующему курсу до ТПН со средним временем от момента появления до ТПН, равным 4.6 лет [52]. Первичные варианты лечения пациентов с терминальной стадией заболевания почек от AFib включают изолированную трансплантацию почки и комбинированную трансплантацию печени и почек [56, 57]. У 10 пациентов, получавших изолированную трансплантацию почки, Pinney et al. сообщил о 5- и 10-летней выживаемости трансплантатов 85 и 30% соответственно. Рецидив амилоида выявлен у семи пациентов. Девяти пациентам была выполнена комбинированная трансплантация печени и почек, при этом выживаемость трансплантата в течение 5 и 10 лет составила 63 и 31% соответственно.Ни у одного пациента в этой группе не развился рецидив амилоидоза [28].
Лизоцим — бактериолитический фермент, который синтезируется гепатоцитами и лейкоцитами, включая нейтрофилы и макрофаги. Вариант лизоцима, приводящий к системному амилоидозу, был впервые описан в 1993 г. и с тех пор был зарегистрирован в семьях английского, французского и итальянского происхождения [58–62]. Описаны по крайней мере четыре патогенные мутации, все локализованные в экзоне 2 гена лизоцима. Отложение амилоида может быть широко распространенным, и пораженные органы включают почки, печень, желудочно-кишечный тракт, селезенку, лимфатические узлы, слюнные железы и кожу.Внепочечные проявления включают желудочно-кишечное кровотечение, мегалобластную анемию из-за мальабсорбции и разрыв печени [59]. Поражение почек описано во многих, но не во всех отчетах об амилоидозе ALys. Почечные проявления варьируются от легкой протеинурии до прогрессирующей почечной недостаточности, требующей заместительной почечной терапии. Биопсия почек показала отложение амилоида во всех отделах; однако клубочки и артериолы, по-видимому, поражаются наиболее часто и серьезно. Сообщалось также об интерстициальных отложениях в коре и мозговом веществе [59, 61, 62].Прогрессирование ALys в ESRD, по-видимому, является медленным процессом. Пинни и др. сообщил о трех пациентах с ALys, которым была проведена трансплантация почек со средним временем прогрессирования до ТПН 10,4 года. У всех трех пациентов на момент публикации была хорошая функция трансплантата без признаков рецидива амилоида [28]. Два пациента в семье, о которых сообщили Yazaki et al. была проведена трансплантация почки, длительное наблюдение показало хорошую функцию [62].
Варианты аполипопротеина AI и AII представляют собой хорошо описанные формы наследственного системного амилоидоза с поражением почек [53].И аполипопротеин AI, и AII синтезируются в печени и тонком кишечнике и являются структурными компонентами ЛПВП [63]. В случае AApo AI было описано по крайней мере 19 мутаций в гене APOAI , приводящих к амилоидозу [64]. Мутации около N-конца связаны с отложениями в почках, печени и иногда в сердце, тогда как мутации около C-конца связаны с отложениями в сердце, на коже и гортани [53, 63, 64]. У мужчин может происходить отложение AApo AI в яичках, и гипогонадизм может быть первым признаком заболевания [65].Заболевание почек при AApo AI часто проявляется как медленно прогрессирующая почечная недостаточность с минимальной протеинурией; однако сообщалось о нефротическом синдроме. Отложения амилоида в почках в основном наблюдались в интерстиции мозгового вещества с сохранением коркового слоя почек [66]. Также описаны два случая AApo AI с более обширным поражением клубочков и сосудов [30, 67]. Впервые об амилоидогенном аполипопротеине AII было сообщено в 2001 году, и все описанные мутации произошли в стоп-кодоне, что привело к удлинению белка дикого типа на 21 аминокислоту [68, 69].Клинически преобладает заболевание почек, и у пациентов обычно наблюдается прогрессирующая почечная недостаточность и протеинурия. Гистологически отложения амилоида AApo AII наиболее часто обнаруживаются в клубочках и мелких сосудах. При вскрытии обнаруживаются внепочечные амилоидные отложения, в основном в стенках сосудов [53, 68]. Недавно был описан случай аполипопротеинового амилоидоза AIV (AApo AIV) с преимущественно медуллярным отложением амилоида [70]. Амилоидоз, вызванный вариантом аполипопротеина AI, AII или AIV, очень редок и в двух исследованиях составлял <1% биопсий амилоида почек [7, 30].Специфического лечения амилоидоза AApo AI, AII или AIV не существует, а трансплантация почки оказалась эффективной в случаях, прогрессирующих до органной недостаточности. Четырнадцать пациентов, получавших либо изолированную почку, либо комбинированную трансплантацию почек-печени или сердца-почки для AApo AI, были включены в исследование Pinney et al. со средней выживаемостью трансплантата 13,1 года. 5- и 10-летняя выживаемость составила 100 и 77% соответственно, при этом только один пациент потерял трансплантат из-за рецидивирующего амилоидоза [28].Имеется немного сообщений о трансплантации почки при амилоидозе AApo AII, но описано долгосрочное выживание трансплантата до 10 лет [71].
Транстиретиновый амилоидоз — наиболее распространенный тип семейного амилоидоза, который в первую очередь связан с периферической невропатией и дизавтономией. Другие участки поражения включают сердце, кожу, желудочно-кишечный тракт и почки. Транстиретин преимущественно синтезируется в печени, и в гене TTR было описано не менее 100 различных мутаций, приводящих к амилоидозу, с до 15 нефропатическими вариантами [72].Вариант TTR Val30Met является наиболее распространенным и распространен по всему миру. Транстиретин дикого типа также может образовывать амилоидные фибриллы и участвует в системном старческом амилоидозе. Возраст начала может быть весьма различным, а семейный анамнез может быть трудно раскрыть из-за позднего начала заболевания [9, 72]. Поражение почек часто сопровождается микроальбуминурией, и в одном исследовании 50% пациентов с микроальбуминурией прогрессировали до явной нефропатии, а 32% из них показали прогрессирование до ТПН [73].При биопсии почки отложения ATTR были идентифицированы во всех почечных компартментах. Внутри клубочка чаще всего поражаются мезангиальные области, за которыми следуют артериолы на сосудистом полюсе [73]. Тубулоинтерстициальные отложения являются частой находкой и часто наиболее заметны в мозговом веществе почек [74, 75]. Долгосрочные результаты у пациентов с прогрессирующей нефропатией плохие: средняя выживаемость 21 месяц после начала диализа в исследовании [76]. Окончательное лечение включает трансплантацию печени, чтобы остановить выработку мутировавшего транстиретина.Результаты после трансплантации печени не всегда благоприятны и, вероятно, связаны с основной степенью отложения амилоида и дисфункцией органа во время трансплантации. Комбинированная трансплантация печени и почек была выполнена ряду пациентов с наследственным ATTR, и в исследовании у шести пациентов, получавших комбинированную трансплантацию, не было выявлено рецидива протеинурии через 84 месяца [77].
Гельсолиновый амилоидоз (AGel) — редкая форма наследственного системного амилоидоза, которая в первую очередь проявляется как синдром аутосомно-доминантной полинейропатии.Кардинальные клинические проявления амилоидоза AGel включают решетчатую дистрофию роговицы, прогрессирующие невропатии и кутис-лаксию [78, 79]. Заболевание почек встречается редко, но о нем сообщалось. У пациентов с гомозиготными мутациями в гене гельсолина почечные проявления, включая протеинурию и прогрессирующую почечную недостаточность, могут быть более серьезными и возникать раньше, чем у пациентов с гетерозиготными мутациями [80]. Сообщалось по крайней мере о трех мутациях, приводящих к варианту белка гельсолина, и о двух из них (Asp187Asn и Gly194Arg) сообщалось с поражением почек [80–83].Результаты биопсии почек у пациентов с амилоидозом AGel включают преимущественно гломерулярные амилоидные отложения [81, 82]. Sethi et al. сообщил о случае амилоидоза AGel с необычными ультраструктурными особенностями, включая параллельную и спиралевидную организацию амилоидных фибрилл [82]. Как и другие формы наследственного амилоидоза, специфической терапии амилоидоза гельзолином не существует. Сообщалось о трансплантации почки со стабильной функцией трансплантата через 3–6 лет после трансплантации [78, 80].
Сводка
Таким образом, амилоидоз почек является необычным заболеванием, на которое приходится около 2% биопсий нативной почки [7, 30].Все формы амилоида имеют общие гистологические и ультраструктурные данные, но состоят из множества различных белков-предшественников. Определение состава амилоидных отложений имеет первостепенное значение, поскольку оно способствует лечению и предсказывает исход и вероятность внепочечного заболевания. Достижения в диагностических методах, включая появление лазерной микродиссекции и протеомного анализа на основе масс-спектроскопии, привели к большей точности идентификации амилоида.
Заявление о конфликте интересов
Не объявлено.
Список литературы
1.Амилоидоз: клинический обзор
,Rheum Dis Clin N Am
,2013
, vol.39
(стр.323
—345
) 2« и др.Белки амилоидных фибрилл: доказательство гомологии с легкими цепями иммуноглобулинов с помощью анализа последовательностей
,Science
,1971
, vol.172
(стр.1150
—1151
) 3,,.Как я лечу амилоидоз: важность точной диагностики и амилоидного типирования
,Кровь
,2012
, т.120
(стр.3206
—3213
) 4« и др.Лазерная микродиссекция и протеомика на основе масс-спектрометрии помогает в диагностике и типировании амилоидоза почек
,Kidney Int
,2012
, vol.82
(стр.226
—234
) 5.Заболевание почек, связанное с амилоидозом
,J Am Soc Nephrol
,2006
, vol.17
(стр.3458
—3471
) 6,.Молекулярные механизмы амилоидоза
,N Engl J Med
,2003
, vol.349
(стр.583
—596
) 7,,, et al.Амилоидоз почек: происхождение и клинико-патологические корреляции 474 недавних случаев
,Clin J Am Soc Nephrol
,2013
, vol.8
(стр.1515
—1523
) 8« и др.Типирование амилоидоза при биопсии почек: диагностические ошибки
,Arch Pathol Lab Med
,2007
, vol.131
(стр.917
—922
) 9« и др.Неверный диагноз наследственного амилоидоза как AL (первичный) амилоидоз
,N Engl J Med
,2002
, vol.346
(стр.1786
—1791
) 10« и др.AL-амилоидоз не диагностируется при биопсии почек
,Nephrol Dial Transplant
,2004
, vol.19
(стр.3050
—3053
) 11« и др.Классификация амилоидоза с помощью лазерной микродиссекции и протеомного анализа на основе масс-спектрометрии в клинических биоптатах
,Кровь
,2009
, vol.114
(стр.4957
—4959
) 12 и др.Два разных типа амилоидных отложений — аполипопротеин A-IV и транстиретин — у пациента с системным амилоидозом
,Lab Invest
,2004
, vol.84
8
(стр.981
—988
) 13.Диспротеинемия и почка
,Adv Anat Pathol
,2004
, vol.11
(стр.49
—63
) 14« и др.Диагностика и характеристика амилоидоза тяжелых и тяжелых / легких цепей почек и их сравнение с амилоидозом легких цепей почек
,Kidney Int
,2013
, vol.83
(стр.463
—470
) 15,,, et al.Распространенность и морфология амилоида, ассоциированного с хемотаксическим фактором 2 лейкоцитов, в биоптатах почек
,Kidney Int
,2010
, vol.77
(стр.816
—819
) 16« и др.Поражение почек при системном амилоидозе: итальянское совместное исследование выживаемости и исходов для почек
,Nephrol Dial Transplant
,2008
, vol.23
(стр.941
—951
) 17« и др.Клинико-патологические корреляции при множественной миеломе: серия случаев из 190 пациентов с биопсией почек
,Am J Kidney Dis
,2012
, vol.59
(стр.786
—794
) 18,,.Амилоидоз легкой цепи иммуноглобулина и почек
,Kidney Int
,2002
, vol.61
(стр.1
—9
) 19« и др.Клинические исходы амилоидоза легкой цепи иммуноглобулина, поражающего почки
,Nephrol Dial Transplant
,2009
, vol.24
(стр.3132
—3137
) 20« и др.Гены вариабельной (V) области легкой цепи иммуноглобулина влияют на клиническую картину и исход при амилоидозе, ассоциированном с легкой цепью (AL)
,Blood
,2003
, vol.101
(стр.3801
—3808
) 21,,.Паттерн фрагментации фибриллярных белков при системном AL-амилоидозе
,J Pathol
,2009
, vol.219
(стр.473
—480
) 22,.Первичный системный амилоидоз: клинико-лабораторные особенности в 474 случаях
,Semin Hematol
,1995
, vol.32
(стр.45
—59
) 23« и др.Клинические особенности пациентов с амилоидозом легкой цепи иммуноглобулина (AL) с сосудисто-ограниченным отложением в почках
,Nephrol Dial Transplant
,2012
, vol.27
(стр.1097
—1101
) 24« и др.Внутритрубчатый амилоидоз
,Kidney Int
,2007
, vol.72
(стр.1282
—1288
) 25« и др.Почечная недостаточность, вызванная сочетанной нефропатией гипсовой повязки, амилоидозом и болезнью отложений легких цепей
,Трансплантат Nephrol Dial
,2010
, vol.25
(стр.1340
—1343
) 26« и др.Трансплантация высоких доз мелфалана и аутологичных стволовых клеток пациентам с амилоидозом AL: 8-летнее исследование
,Ann Int Med
,2004
, vol.140
(стр.85
—93
) 27,,, et al.Тяжесть исходной протеинурии позволяет прогнозировать почечный ответ при амилоидозе, ассоциированном с легкой цепью иммуноглобулинов, после трансплантации аутологичных стволовых клеток
,Clin J Am Soc Nephrol
,2007
, vol.2
(стр.440
—444
) 28« и др.Трансплантация почки при системном амилоидозе — важность типа амилоидных фибрилл и обилия белков-предшественников
,Am J Transplant
,2013
, vol.13
(стр.433
—441
) 29,,, et al.Клинические и гистологические характеристики почечного амилоидоза AA: ретроспективное исследование 68 случаев с особым интересом к воспалительной реакции, связанной с амилоидом
,Hum Pathol
,2007
, vol.38
(стр.1798
—1809
) 30« и др.Распространенность и происхождение амилоида в биопсиях почек
,Am J Surg Pathol
,2009
, vol.33
(стр.1198
—1205
) 31« и др.Патогенетические механизмы амилоидного амилоидоза А
,P Natl Acad Sci USA
,2013
, vol.110
(стр.16115
—16120
) 32,,.Экспрессия и функция сывороточного амилоида А, основного белка острой фазы, при нормальных и болезненных состояниях
,Curr Opin Hematol
,2000
, vol.7
(стр.64
—69
) 33« и др.Естественное течение и исход при системном амилоидозе AA
,N Engl J Med
,2007
, vol.356
(стр.2361
—2371
) 34« и др.Уникентрическая болезнь Кастлемана, осложненная системным амилоидозом АА: излечимая болезнь
,QJM
,2002
, vol.95
(стр.211
—218
) 35,,, et al.AA амилоидоз, осложняющий наследственные синдромы периодической лихорадки
,Arthritis Rheum
,2013
, vol.65
(стр.1116
—1121
) 36« и др.AA амилоидоз, ассоциированный с гепатитом B
,Nephrol Dial Transplant
,2011
, vol.26
(стр.2407
—2412
) 37« и др.Почечный амилоидоз после туберкулеза
,Indian J Pediatr
,2000
, vol.67
(стр.679
—681
) 38« и др.Рецидивирующий амилоидоз AA в трансплантате почки
,Am J Kidney Dis
,2011
, vol.57
(стр.941
—944
) 39« и др.AA амилоидоз, связанный с мутировавшим сывороточным амилоидным белком A4
,Amyloid
,2009
, vol.16
(стр.84
—88
) 40,,.Амилоидоз почек у детей
,Педиатр Нефрол
,2011
, т.26
(стр.1215
—1227
) 41« и др.Сравнение клинической применимости тоцилизумаба и терапии анти-TNF при амилоидозе AA, осложняющем ревматические заболевания
,Mod Rheumatol
,2014
, vol.24
(стр.137
—143
) 42,,, et al.Эпродизат для лечения почечной недостаточности при амилоидозе AA
,N Engl J Med
,2007
, vol.356
(стр.2349
—2360
) 43« и др.Хемотаксический фактор лейкоцитов 2: новый почечный амилоидный белок
,Kidney Int
,2008
, vol.74
(стр.218
—222
) 44« и др.Молекулярное клонирование, структурная характеристика и хромосомное картирование гена LECT2 человека
,Genomics
,1998
, vol.48
(стр.324
—329
) 45,,, et al.Системная экспрессия недавно обнаруженного белка LECT2 в организме человека
,Pathol Int
,1998
, vol.48
(стр.882
—886
) 46« и др.Почечный амилоидоз, связанный с лейкоцитарным хемотаксическим фактором 2 (LECT2): серия случаев
,Am J Kidney Dis
,2010
, vol.56
(стр.1100
—1107
) 47« и др.Почечный ALECT2 амилоидоз: клинико-патологическое исследование 17 пациентов
,Mod Pathol
,2013
, vol.26
Доп. 2
стр.392A
48« и др.Клинический и патологический фенотип лейкоцитарного клеточного хемотаксин-2 (LECT2) амилоидоза (ALECT2) [аннотация OP-058]
,Amyloid
,2010
, vol.17
доп. 1
(стр.69
—70
) 49,,, et al.Амилоидоз лейкоцитарного хемотаксического фактора 2 (LECT2), проявляющийся как легочно-почечный синдром: описание случая и обзор литературы
,Clin Kidney J
,2013
, vol.6
(стр.618
—621
) 50« и др.Атипичное представление атипичного амилоида
,Циферблат нефрола
,2011
, т.26
(стр.373
—376
) 51,,, et al.Наследственный амилоидоз почек, связанный с альфа-цепью мутантного фибриногена
,Nat Gen
,1993
, vol.3
(стр.252
—255
) 52« и др.Диагностика, патогенез, лечение и прогноз наследственного амилоидоза альфа-цепи фибриногена А
,J Am Soc Nephrol
,2009
, vol.20
(стр.444
—451
) 53.Остертаг снова: унаследованные системные амилоидозы без нейропатии
,Амилоид
,2005
, т.12
(стр.75
—87
) 54« и др.Рецидив амилоидоза при трансплантации почки
,Am J Kidney Dis
,2010
, vol.56
(стр.394
—398
) 55,,.Впервые возникшая протеинурия с массивными аморфными клубочковыми отложениями
,Am J Kidney Dis
,2010
, vol.55
(стр.749
—754
) 56« и др.Успешная трансплантация печени при наследственном амилоидозе, вызванном мутацией сдвига рамки считывания в гене альфа-цепи фибриногена
,Am J Transplant
,2006
, vol.6
(стр.632
—635
) 57« и др.Ортотопическая трансплантация печени при наследственном фибриноген-амилоидозе
,Трансплантация
,2003
, т.75
(стр.560
—561
) 58« и др.Мутации гена лизоцима человека вызывают наследственный системный амилоидоз
,Nature
,1993
, vol.362
(стр.553
—557
) 59« и др.Лизоцимный амилоидоз: отчет о 4 случаях и обзор литературы
,Медицина
,2006
, vol.85
(стр.66
—73
) 60« и др.Наследственный амилоидоз почек, связанный с вариантным лизоцимом в большой английской семье
,Nephrol Dial Transplant
,1999
, vol.14
(стр.2639
—2644
) 61« и др.Наследственный амилоидоз почек, вызванный новым вариантом лизоцима W64R во французской семье
,Kidney Int
,2002
, vol.61
(стр.907
—912
) 62,,.Новая мутация лизоцима Phe57Ile, связанная с наследственным амилоидозом почек
,Kidney Int
,2003
, vol.63
(стр.1652
—1657
) 63« и др.Структура, функция и амилоидогенная предрасположенность аполипопротеина A-I
,Амилоид
,2006
, т.13
(стр.191
—205
) 64« и др.Амилоидогенность и клинический фенотип, связанный с пятью новыми мутациями аполипопротеина A-I
,Am J Pathol
,2011
, vol.179
(стр.1978
—1987
) 65,,, et al.Бесплодие и гипергонадотропный гипогонадизм как первые свидетельства наследственного аполипопротеинового амилоидоза A-I
,J Urology
,2007
, vol.178
(стр.344
—348
) 66« и др.Почечный аполипопротеиновый амилоидоз A-I: редкая и обычно игнорируемая причина наследственного тубулоинтерстициального нефрита
,J Am Soc Nephrol
,2005
, vol.16
(стр.3680
—3686
) 67« и др.Почечный аполипопротеиновый амилоидоз A-I, связанный с новым мутантом Leu64Pro
,Am J Kidney Dis
,2004
, vol.44
(стр.1103
—1109
) 68« и др.Наследственный системный амилоидоз, связанный с новой мутацией стоп-кодона аполипопротеина AII Stop78Arg
,Kidney Int
,2003
, vol.64
(стр.11
—16
) 69« и др.Новый наследственный амилоидоз человека: результат мутации стоп-кодона в гене аполипопротеина AII
,Genomics
,2001
, vol.72
(стр.272
—277
) 70,,, et al.Медуллярный амилоидоз, связанный с отложением аполипопротеина A-IV
,Kidney Int
,2012
, vol.81
(стр.201
—206
) 71« и др.Трансплантация почки при амилоидозе аполипопротеина AII
,Амилоид
,2003
, т.10
(стр.224
—228
) 72,.Транстиретиновый амилоидоз и почки
,Clin J Am Soc Nephrol
,2012
, vol.7
(стр.1337
—1346
) 73« и др.Семейный амилоидоз ATTR: микроальбуминурия как прогностический фактор симптоматического заболевания и клинической нефропатии
,Nephrol Dial Transplant
,2003
, vol.18
(стр.532
—538
) 74« и др.Семейная амилоидная полинейропатия I типа (португальский язык): распределение и характеристика отложений амилоида в почках
,Am J Kidney Dis
,1998
, vol.31
(стр.940
—946
) 75,,.Значение биопсии почек в прогнозе трансплантации печени при семейной амилоидной полинейропатии Пациенты ATTR Val30Met
,Amyloid
,2006
, vol.13
(стр.99
—107
) 76« и др.Терминальная стадия почечной недостаточности и диализ при наследственном амилоидозе TTR V30M: презентация, выживаемость и прогностические факторы
,Amyloid
,2004
, vol.11
(стр.27
—37
) 77« и др.Комбинированная трансплантация печени и почек при семейной амилоидотической полинейропатии TTR V30M: нефрологическая оценка
,Amyloid
,2011
, vol.18
Дополнение 1
(стр.190
—192
) 78,,, et al.Исход трансплантации почки при наследственном гельсолин-амилоидозе
,Am J Med Sci
,2009
, vol.337
(стр.370
—372
) 79.Семейный системный парамилоидоз с решетчатой дистрофией роговицы, прогрессирующей черепной невропатией, кожными изменениями и различными внутренними симптомами. Ранее нераспознанный наследственный синдром
,Ann Clin Res
,1969
, vol.1
(стр.314
—324
) 80,,, et al.Гомозиготность по мутации гельсолина Asn187 при семейном амилоидозе финского типа связана с тяжелым заболеванием почек
,Genomics
,1992
, vol.13
(стр.902
—903
) 81,,.Нефротический синдром, связанный с амилоидозом, вызванный мутацией гельсолина G654A: первое сообщение с Ближнего Востока
,Nephrol Dial Transplant
,2007
, vol.22
(стр.272
—275
) 82« и др.Почечный амилоидоз, связанный с новым вариантом последовательности гельсолина
,Am J Kidney Dis
,2013
, vol.61
(стр.161
—166
) 83« и др.Гельзолин-производный семейный амилоидоз, вызванный аспарагиновой или тирозиновой заменой аспарагиновой кислоты по остатку 187
,Nat Gen
,1992
, vol.2
(стр.157
—160
)© Автор, 2014. Опубликовано Oxford University Press от имени ERA-EDTA. Все права защищены. Для получения разрешений обращайтесь по электронной почте: [email protected].
Это статья в открытом доступе, распространяемая в соответствии с условиями некоммерческой лицензии Creative Commons Attribution (http: // creativecommons.org / licenses / by-nc / 4.0 /), который разрешает некоммерческое повторное использование, распространение и воспроизведение на любом носителе при условии правильного цитирования оригинальной работы. По вопросам коммерческого повторного использования обращайтесь по адресу [email protected]
. .