Лечение мышечной дистрофии дюшенна. Список клиник, рейтинг, отзывы, цены
О заболевании
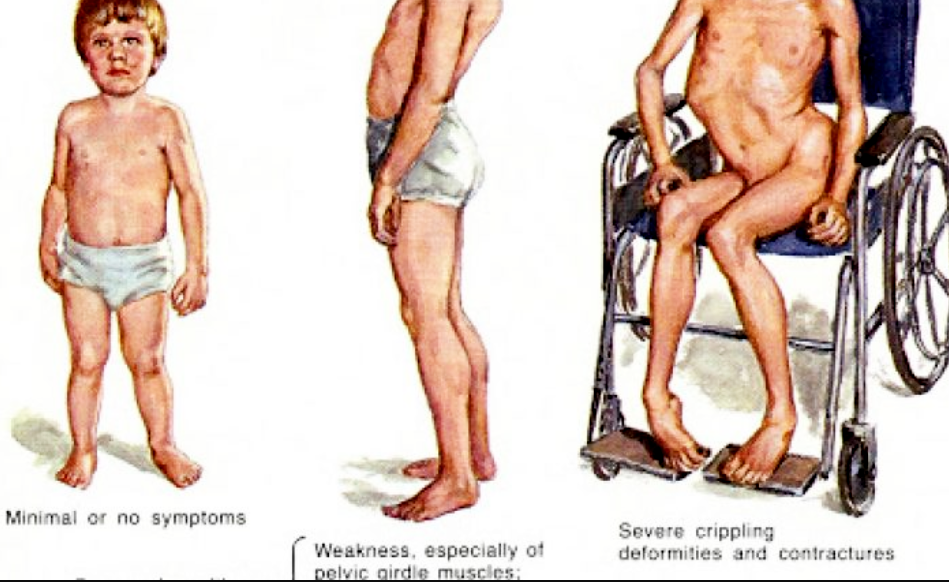
Принадлежащая к категории редких заболеваний мышечная дистрофия Дюшенна имеет врожденный характер. Этот избирательный недуг поражает только представителей мужского пола. Для него свойственна мышечная слабость, которая является следствием постепенного разрушения связей на нервно-мышечном уровне. Передается данный недуг по наследству, поэтому носителями гена могут быть женщины, хотя сами не подвержены заболеванию. Период активного развития – до десяти лет. Ранние симптомы можно заметить еще в младенческом возраcте. Такие дети плохо сосут грудь, а когда они взрослеют, им не удается сложить губы для свиста. Также весьма проблематично поднимание рук выше головы. При выражении эмоций их лица не изменяются, а при плаче и смехе мимика кардинально отличается от обычной.
Синдром Дюшенна передается по наследству, имея модифицированный ген, приводящий к сбоям в организме. В результате модификации гена, отвечающего за выработку протеина, возникают серьезные проблемы с образованием полноценной мышечной ткани. В результате современных исследований удалось установить, что недуг может быть не только врожденным, он может появиться у мальчиков, в генах которых произошли мутации уже после рождения.
В результате современных исследований удалось установить, что недуг может быть не только врожденным, он может появиться у мальчиков, в генах которых произошли мутации уже после рождения.
Самые начальные признаки проявляются до пяти лет. У ребенка, который заболел после рождения, наблюдается постепенное торможение двигательных функций. Постепенно происходи увеличение объемов определенных групп мышц, что внешне выглядеть как сильно накачанная мускулатура, которая постепенно истощается. Распространение болезни происходит снизу-вверх, от мышц ног – к спине и рукам.
Симптомы
Синдром Дюшенна начинает проявлять себя к восемнадцати месяцам. Ребенок не может начать самостоятельно ходить, все его попытки стать самостоятельно на ноги и сделать шаги безуспешны. Ребенок долго не может начать ходить, серьезно опаздывая и отставая от ровесников. В последующем он часто падает и ходит раскачиваясь. Одним из признаков заболевания является положение ребенка, который, желая встать, руками поддерживает ноги. Чрезвычайно слабыми являются все мышцы, которые не могут выполнять свои функции. Эти и другие признаки позволят врачу заподозрить недуг, чтобы направить ребенка на консультацию к более узкому специалисту – ортопеду.
Чрезвычайно слабыми являются все мышцы, которые не могут выполнять свои функции. Эти и другие признаки позволят врачу заподозрить недуг, чтобы направить ребенка на консультацию к более узкому специалисту – ортопеду.
Диагностика
- Для подтверждения диагноза понадобится анализ крови, проведения теста, определяющего состояние мышц. Для этой цели используется электромиографическая методика, посредством которой измеряется скорость проведения нервных импульсов в мышцах. Данные полученные в результате обследования больных детей резко отличаются от нормы. Также прибегают к биопсии мышц. Под микроскопом обнаруживаются изменения на клеточном уровне, а также удается рассмотреть отложения жировой ткани.
- Обязательно проводится оценка сердечной деятельности и работы дыхательной системы, мышечных функций. Проводится ЭКГ, также используется методика определения сердечных маркеров и плотности костной ткани. Точный диагноз необходимо поставить как можно раньше, чтобы получить четкую картину заболевания и правильно информировать родителей об особенностях его протекания.


Виды лечения
- Лечения синдрома Дюшенна за рубежом и в нашей стране еще не существует. Но сегодня ведутся постоянные исследования, которые направлены на поиск эффективных средств, которые бы могли облегчить симптомы и снизить темп прогрессирования. Для этого используют методику введения гена дистрофина, что позволяет сохранить двигательные функции. Также прибегают к имплантации клеток, которые усиливают синтез дистрофина, не подвергающегося мутации. Применение данной технологии дает стойкий результат, который заметен в любом возрасте. Используются также возможности гена атрофина. В настоящее время специалисты назначают препараты, направленные на увеличение мышечных функций, что значительно облегчает состояние пациента. Этой цели также помогают достичь физиотерапевтические процедуры. Больному также понадобится ортопедическая помощь – коляски, ходунки, фиксаторы голени.
Автор: Доктор Надежда Иванисова
Эффективные методики лечения дистрофии Дюшенна: существуют ли они?
«Я не смогу это пережить. ..», — первая мысль, которая приходит в голову матери, узнавшей подробности о болезни ее ребенка. До постановки диагноза большинство даже не знает, что это за заболевание — миопатия Дюшенна (также известна как дистрофия и миодистрофия Дюшенна). И неудивительно, ведь частота встречаемости достаточно низкая: всего 1 случай на 3500 рождений мальчиков. Крайне редко эта генетическая болезнь встречается и у девочек. Но что вам до редкости заболевания, когда в карточке вашего малыша появляется запись, которую вам хотелось бы бесследно стереть?
..», — первая мысль, которая приходит в голову матери, узнавшей подробности о болезни ее ребенка. До постановки диагноза большинство даже не знает, что это за заболевание — миопатия Дюшенна (также известна как дистрофия и миодистрофия Дюшенна). И неудивительно, ведь частота встречаемости достаточно низкая: всего 1 случай на 3500 рождений мальчиков. Крайне редко эта генетическая болезнь встречается и у девочек. Но что вам до редкости заболевания, когда в карточке вашего малыша появляется запись, которую вам хотелось бы бесследно стереть?
Не упустить время
Милая неуклюжесть, сопровождающая малыша до трех лет, к пяти превращается в мышечную слабость. В 10-12 лет ребенок перестает ходить, к 15 — частично или полностью теряет способность двигаться самостоятельно. Всего 20 лет жизни — так мало, что в это невозможно поверить.
Врачи отделываются общими фразами, и родителям приходится искать информацию самостоятельно. Доктора, впервые столкнувшиеся с таким диагнозом, и сами зачастую не представляют, что делать.
Миопатия Дюшенна — заболевание, которое характеризуется мутацией в гене, отвечающем за синтез белка дистрофин в мышечных волокнах, что приводит к нарушению их строения. Носители мутаций — женщины, которые сами миопатией Дюшенна не болеют, но с 50% вероятностью передают ее своим потомкам. У мальчиков при этом развивается мышечная дистрофия, а девочки, в свою очередь, становятся носителями мутации. В некоторых случаях заболевание не является результатом передачи по наследству, а возникает спонтанно.
В России гигантский провал в данной области научных исследований. Специалистов по нейромышечным патологиям крайне мало, попасть к ним сложно. Только представьте, как много значит один год для прогрессирующего заболевания! А именно столько в среднем длится очередь в федеральное медучреждение.
Обычно сначала родители из-за трудностей при ходьбе обращаются к ортопедам, которые могут просто не знать об этом заболевании. Как показывает практика российских поликлиник, даже неврологи во многих случаях ошибаются с диагнозом.
Судя по частоте заболевания, в России должно быть около 4000 детей с дистрофией Дюшенна. На учете в московском детском нервно-мышечном центре — в 10 раз меньше. Где все остальные пациенты?
В этом-то и кроется самая большая проблема отечественной медицины. Невролог, наконец-то правильно поставивший диагноз, вольно или невольно убеждает родителей, что миопатия Дюшенна не лечится, быстро прогрессирует и всегда приводит к смерти. В результате — родители просто опускают руки, никуда не едут и ни к кому не обращаются, пытаясь просто выжить в поставленных условиях.
Правда ли, что ситуация настолько безвыходна? Неужели нет надежды на то, что ребенку действительно помогут? Разве неизлечимое заболевание — это повод отказаться от поисков наиболее эффективного лечения?
Учиться и работать с дистрофией Дюшенна — реально
Будем честны. Пока чуда мгновенного излечения ждать не стоит, кто бы вам что ни обещал. Однако продлить жизнь ребенка и в разы улучшить ее качество — возможно уже сейчас! Велик шанс, что за те годы, что вы ему подарите, ученые наконец-то найдут средство остановить или повернуть вспять развитие заболевания.
Пока чуда мгновенного излечения ждать не стоит, кто бы вам что ни обещал. Однако продлить жизнь ребенка и в разы улучшить ее качество — возможно уже сейчас! Велик шанс, что за те годы, что вы ему подарите, ученые наконец-то найдут средство остановить или повернуть вспять развитие заболевания.
Центр комплексного лечения нейромышечных нарушений Детской больницы Цинциннати (Comprehensive Neuromuscular Center, Цинциннати, США) доказывает это каждый день:
-
Здесь регулярно проводят клинические испытания новых лекарств и методов, что позволило достичь отличных результатов в лечении мышечной миопатии Дюшенна.
-
На данный момент в Центре наблюдаются около 600 детей с этим заболеванием. Сюда приезжают со всего мира, чтобы получить современное лечение и эмоциональную поддержку. Медицинская команда оказывает своим пациентам и их семьям долгосрочную помощь.
-
Наблюдения за пациентами 13-16 лет, которые выполняли все назначения врачей Центра, подтверждают: к этому возрасту 40% из них могут самостоятельно встать с пола, 50% — пройти 10 метров без посторонней помощи.
Тогда как без лечения ходьба становится абсолютно невозможной уже к 12-13 годам.
 Тогда как без лечения ходьба становится абсолютно невозможной уже к 12-13 годам.
Тогда как без лечения ходьба становится абсолютно невозможной уже к 12-13 годам.-
Все специалисты собраны в одном учреждении. Совместная работа неврологов, кардиологов, пульмонологов, эндокринологов, генетиков, физиотерапевтов, диетологов приводит к тому, что средняя продолжительность жизни людей с миопатией Дюшенна увеличилась на 10 лет.
Благодаря комплексному лечению и постоянному наблюдению вместо 20 лет пациенты могут жить 30 и более. Соответственно, и основные двигательные функции тоже сохраняются намного дольше.
А это значит, что дети могут успешно получить профессиональное образование и даже работать. Такие примеры среди пациентов Центра уже есть!
Эффективные методики лечения миопатии Дюшенна от Comprehensive Neuromuscular Center
Центр комплексного лечения нейромышечных нарушений работает по продуманной схеме, которая делает лечение максимально комфортным для пациента и его законных представителей (родителей или опекунов).
Преимущества лечения миодистрофии Дюшенна в Comprehensive Neuromuscular Center:
- Специализация на нейромышечных нарушениях. В Центр обращаются пациенты с миопатиями Дюшенна и Беккера, спинальной мышечной атрофией (СМА), врожденным миастеническим синдромом, наследственной атаксией Фридрейха, другими патологиями нервной и мышечной системы.
- Пациенты Центра первыми получают максимальный доступ к новым научным открытиям и достижениям, к самым последним методикам и фармацевтическим средствам. А еще — участвуют в клинических исследованиях.
- По результатам обследования лечащий врач составляет индивидуальный план в зависимости от текущего состояния ребенка и сопутствующих заболеваний. Учитывается даже образ жизни семьи в целом — все для того, чтобы предоставить наилучшую помощь.
- Управление побочными действиями. Состояние ребенка постоянно контролируется с тем, чтобы своевременно корректировать количество и вид принимаемых препаратов.
- Всесторонний амбулаторный и стационарный уход, которые выражаются в постоянной заботе. 100% ведение пациента в течение всей жизни вплоть до консультирования по вопросам качества жизни, обучения и адаптации в обществе.
Лечение миодистрофии Дюшенна будет намного результативнее, если в семье поддерживается благоприятная психологическая обстановка. Поэтому в заключение мы подготовили несколько советов, которые помогут вам сохранять спокойствие и уверенность, а ребенку — стать счастливее.
- Не скрывайте от ребенка диагноз, правдиво и понятно рассказывайте ему о заболевании.
- Помогите вашему ребенку понять, что он может и должен быть активным участником процесса лечения. Правильно питаясь и выполняя рекомендованные физические упражнения, пациент и сам положительно влияет на ход течения болезни.
- Помните, что ваш ребенок — это личность, состоящая из многих аспектов. И миопатия Дюшенна — лишь один из них, причем не самый главный.
- Обращайте внимание на то, что ребенок еще может сделать. Не зацикливайтесь на потерянных умениях.
- Решайте только актуальные проблемы. Не думайте о том, как ребенок будет чувствовать себя через месяц или через год.
- Воспитывайте ребенка так же, как здорового. Старайтесь избежать чрезмерной опеки, предоставьте ему право быть независимым в доступных для него сферах.
- Просите о помощи близких, друзей, врачей тогда, когда вам это нужно.
- По возможности ходите вместе в кино, ездите в отпуска, развлекайтесь — дарите себе и ребенку радостные моменты, наполненные позитивными эмоциями.
- Нужна подробная консультация по нейромышечным нарушениям? Отправьте запрос прямо с сайта и узнайте, как связаться со специалистами Центра комплексного лечения нейромышечных нарушений Детской больницы Цинциннати.

Автор Ольга Тонкушина
Миодистрофия Дюшенна: лучший российский и мировой опыт для российских врачей и пациентского сообщества
На первой всероссийской врачебно-пациентской конференции по миодистрофии Дюшенна, что состоялась 11 декабря, участники обсудили диагностику, реабилитацию, ведение и лечение заболевания, а также сравнили российские и мировые практики.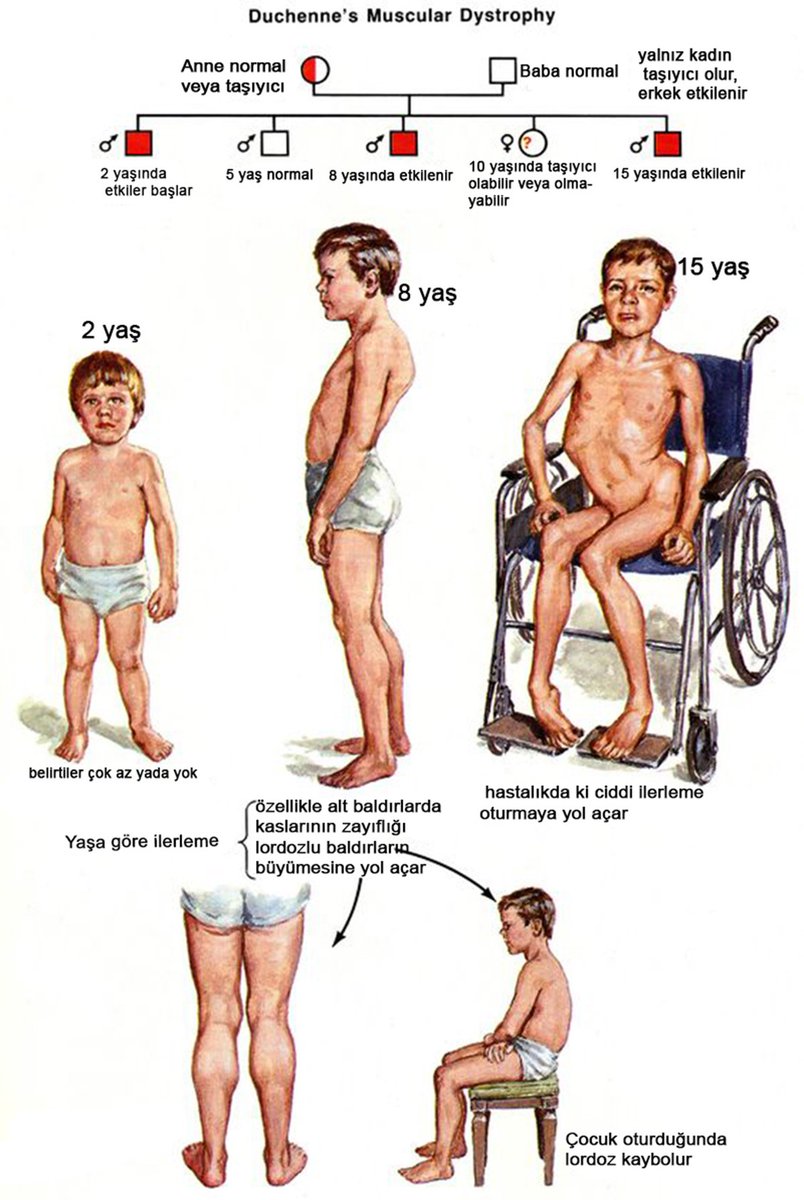
Всего на мероприятие зарегистрировались порядка 500 человек из России, Белоруссии, Украины, Казахстана и Молдавии, среди них – 44% врачей, 48% — родителей пациентов, порядка 5% — представителей некоммерческих организаций. Конференция, организованная молодым благотворительным фондом «Гордей» (он создан в начале 2020 г.), привлекла медицинских экспертов из Италии, США, Нидерландов, Бразилии и Австралии.
— Мы пригласили генетиков, клиницистов и представителей пациентских сообществ ко всестороннему обсуждению проблем диагностики миодистрофии Дюшенна. Врач может увидеть заболевание в раннем возрасте, именно поэтому важна врачебная настороженность, понимание того, на что следует обращать внимание, какие особенности развития мальчиков могут оказаться симптомами этой пока еще фатальной болезни. Мы бы хотели вместе пациентами, генетиками, педиатрами, неврологами и инфекционистами в следующем 2021 году достучаться до каждого детского врача, прийти в каждый детский сад, чтобы не пропустить малышей с этим заболеванием. Сегодня существуют стандарты ухода, позволяющие предупредить развитие тяжелых осложнений болезни, продлить стадию функциональной активности. Кроме того, есть перспектива получения патогенетического лечения и возможность снижения рисков рождения последующих детей с миодистрофией Дюшенна в семьях, — отмечает президент и медицинский директор благотворительного фонда «Гордей», доктор медицинских наук Татьяна Гремякова.
Сегодня существуют стандарты ухода, позволяющие предупредить развитие тяжелых осложнений болезни, продлить стадию функциональной активности. Кроме того, есть перспектива получения патогенетического лечения и возможность снижения рисков рождения последующих детей с миодистрофией Дюшенна в семьях, — отмечает президент и медицинский директор благотворительного фонда «Гордей», доктор медицинских наук Татьяна Гремякова.
В последние несколько лет ситуация с лечением редких генетических заболеваний в РФ меняется к лучшему. Организаторы конференции надеются, что мероприятие поможет привнести в российскую медицинскую практику и семьи международные стандарты ухода и ведения мышечной дистрофии Дюшенна.
— В Италии, с улучшением качества ухода и ведения мальчиков на амбулаторной стадии, продлением способности ходить до подросткового возраста, отпала необходимость в проведении пациентам тяжелых операций на голеностопных суставах и позвоночнике, которые не делаются уже более десяти лет, — рассказал профессор, невролог Марчелло Вилланова (Италия).
Отметим, что в текущем году миодистрофии Дюшенна уделялось повышенное внимание. Как считают эксперты, в диагностике, ведении и лечении этой нозологии в течение одного-двух лет произойдут радикальные перемены. Этому поможет массированное обучение врачей всех специальностей и возможность выявления заболевания в раннем возрасте. И многое уже делается. В 2020 году в России состоялось более 20 конференций, симпозиумов, школ и вебинаров, посвященных различным аспектам заболевания.
— Два наиболее распространенных среди редких и тяжелых по течению генетических детских нейромышечных заболевания – миодистрофия Дюшенна и спинальная мышечная атрофия – составляют 90%. Если педиатры и детские неврологи научатся распознавать их в досимптоматическом периоде, это позволит обеспечить надлежащий уход и даст возможность эффективно применять патогенетичекую терапию, радикально изменив к лучшему жизнь детей, — отметил профессор, детский невролог Лоран Серве (Великобритания).
24 ноября этого года в России зарегистрирован первый таргетный патогенетический препарат для лечения миодистрофии Дюшенна компании – Аталурен компании PTC Therapeutics.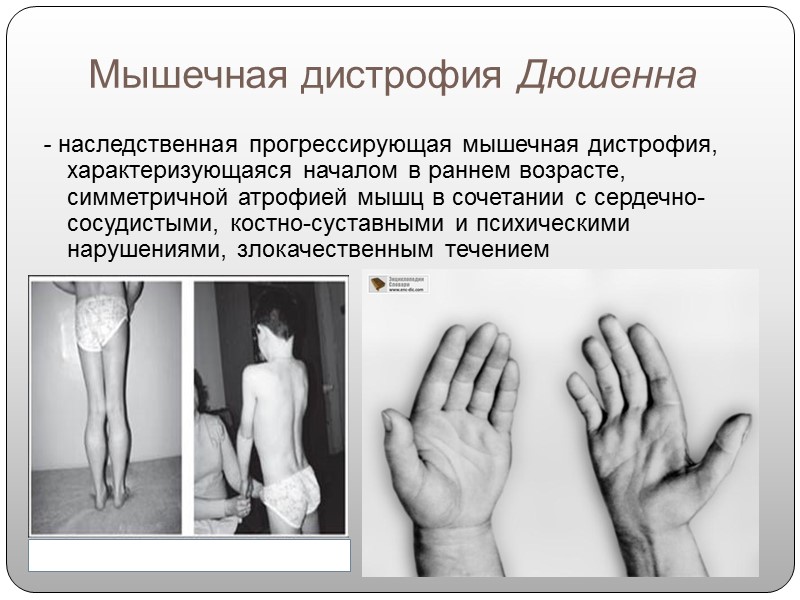 В Общественной палате РФ состоялась серия заседаний по формированию Фонда финансирования лечения для пациентов с орфанными заболеваниями. Фонд создан и, по информации, обнародованной 8 декабря, миодистрофия Дюшенна вошла в число финансируемых нозологий. Аталурен также вошел в список лекарств, на которые были выделены бюджетные средства для больных с нонсенс-мутациями.
В Общественной палате РФ состоялась серия заседаний по формированию Фонда финансирования лечения для пациентов с орфанными заболеваниями. Фонд создан и, по информации, обнародованной 8 декабря, миодистрофия Дюшенна вошла в число финансируемых нозологий. Аталурен также вошел в список лекарств, на которые были выделены бюджетные средства для больных с нонсенс-мутациями.
Для справки: Мышечная дистрофия Дюшенна — редкое генетическое заболевание, поражающее одного мальчика из 3500-5000 новорожденных. Первые признаки и симптомы часто проявляются в возрасте 2-3 лет. Данное заболевание вызвано генетической мутацией, которая не позволяет организму вырабатывать белок дистрофин, стабилизирующий работу мышц. Без него мышечные клетки повреждаются и разрушаются, пациенты со временем утрачивают способность самостоятельно передвигаться, а в дальнейшем могут развиваться сердечная и дыхательная недостаточность.
Мышечная дистрофия | Неврология | Заболевания
Мышечная дистрофия – это патологическое заболевание, которое характерно для людей, ведущих лежачий образ жизни. Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Виды
Мышечная дистрофия очень распространенное патологическое заболевание. Бывает детская и взрослая дистрофия мышц. Также мышечная дистрофия имеет наследственный характер (генетическая и наследственная дистрофия). По характеру и месту локализации различают:
-
инфекционную и неинфекционную;
-
миотоническую;
-
тазово-плечевую;
-
врожденную;
-
плечелопаточную;
.
Симптомы
Мышечная дистрофия прогрессирует заболевание мышечной слабости и потери трудоспособности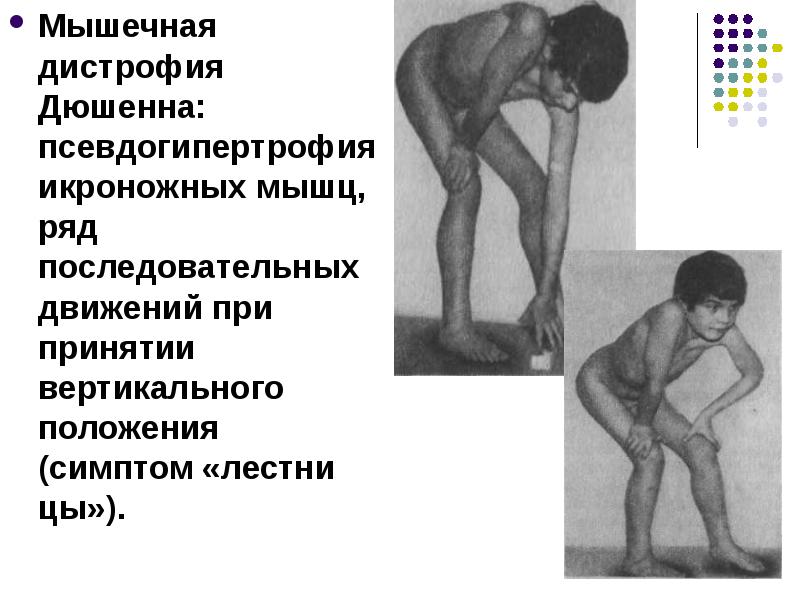 Мышечная дистрофия прогрессирующее заболевание. Поэтому не следует откладывать визит к доктору.
Мышечная дистрофия прогрессирующее заболевание. Поэтому не следует откладывать визит к доктору.
Диагностика
Обратиться к доктору следует немедленно, как только вы заметили мышечную слабость.. К методам диагностики на данном этапе развития медицины относят МРТ. Оно покажет анатомические и физиологические изменения в организме. При этом заболевании также стоит сдать общий анализ крови, мочи и кала. После проведенных диагностик доктор поставит диагноз и направит на лечение.
Лечение
Лечение проводиться с помощью комплексной терапии: консервативное лечение и физиотерапия. Еще не разработано лечение, которые бы полностью устранило это заболевание. Консервативное лечение составляет прием кортикостероидов и препаратов для улучшения мышечной массы. Физиотерапия очень распространенный метод.
Профилактика
Лечение мышечной дистрофии продолжается его профилактикой. Очень важно после выписки с госпиталя не забыть о приписках врача. Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
Найден новый способ лечения мышечной дистрофии Дюшенна
Светлана МасловаФото: US Department of Health and Human ServicesЭкспериментальное лечение не использует методы генной терапии. Оно оказалось эффективным для мышей. Перспективность терапии для человека очень многообещающая, заявили ученые.
382
Ученые из Пенсильванского университета определили группу молекул, которые открывают возможности для разработки лечения людей с мышечной дистрофии Дюшенна — генетическим заболеванием, при котором мышечные волокна испытывают недостаток белка дистрофина. Ученые стремились найти методы лечения болезни не прибегая к генной терапии. Первые результаты доказывают, что это возможно.
Сегодня основным направлением в работе многих групп ученых является увеличение белка утрофина, поскольку он имеет схожие с дистрофином характеристики и генетическую структуру.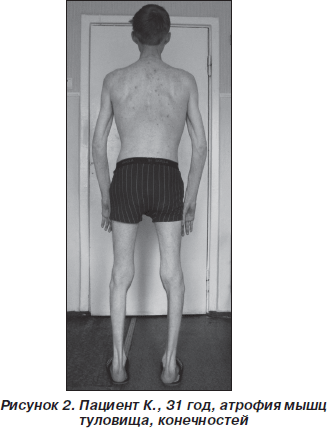 Введение дистрофина с помощью генной терапии связано с рядом ограничений и потенциальных побочных эффектов, поэтому некоторые ученые работают с альтернативными вариантами.
Введение дистрофина с помощью генной терапии связано с рядом ограничений и потенциальных побочных эффектов, поэтому некоторые ученые работают с альтернативными вариантами.
Организм человека с мышечной дистрофией Дюшенна не вырабатывает дистрофин, однако продуцирует утрофин, который потенциально может его стать заменителем.
В экспериментах с мышами ученые протестировали 10 молекул, из которых впоследствии выделили одну — трихостатин А. Терапия трихостатином А привела к значительному улучшению функций и структуры мышечных волокон, показали эксперименты.
«Это совершенно новый метод увеличения утрофина для терапии мышечной дистрофии Дюшенна. Мы еще в начале пути, однако перспективы лечения открывают большие возможности для лечения человека», — заявил старший автор исследования Тейвир Хурана.
На сегодняшний день болезнь неизлечима и уже с подросткового возраста приковывает пациентов к инвалидному креслу. В большинстве случаев заболевание значительно сокращает продолжительность жизни.
Перспективные результаты в лечении мышечной дистрофии Дюшенна недавно представили ученые из США и Канады. Они работают с генетическими инструментами против болезни — коктейлем из молекул ДНК и технологией CRISPR.
Facebook38Вконтакте2WhatsAppTelegram
Препарат для борьбы с мышечной дистрофией Дюшенна улучшает работу энергетических станций клетки
Мышечная дистрофия Дюшенна – наиболее частое нервно-мышечное наследственное заболевание человека. Частота встречаемости составляет 1 на 3500–5000 новорожденных мальчиков в мире. У болезнь ярко выраженный прогрессирующий характер, и уже в подростковом возрасте человек теряет способность к самостоятельному передвижению, а после возникает сердечная и дыхательная недостаточность. С этим заболеванием живут совсем недолго: большинство пациентов умирают в 15–25 лет.
Причина этой тяжелой патологии кроется в мутации гена белка дистрофина. Он отвечает за связь мышечных волокон с клеточным каркасом. Это позволяет поддерживать целостную структуру и функциональность мускулатуры. Вызванный мутациями дефицит этого белка делает мышечные клетки и волокна крайне нестабильными и восприимчивыми к повреждениям. Хотя точные механизмы развития этой патологии все еще требуют дополнительных исследований, уже известно, что большую роль в потере рабочей активности мышечных клеток играет нарушение работы митохондрий. Ранее авторы статьи выяснили, что при болезни Дюшенна в скелетных мышцах появляются серьезные нарушения: митохондрии, производящие энергию и регулирующие транспорт ионов кальция в клетке, начинают работать гораздо менее эффективно. В то же время отмечается, что благодаря митохондриям, клетки сердца, напротив, способны на ранних этапах заболевания сдерживать развитие неблагоприятного сценария. В своей новой работе биологи из республики Марий Эл совместно с коллегами из подмосковного наукограда Пущино изучили влияние терапевтического агента дефлазакорта на работу митохондрий скелетных мышц мышей с точечной мутацией в гене дистрофина, а также здоровых животных, использованных в качестве контроля.
Это позволяет поддерживать целостную структуру и функциональность мускулатуры. Вызванный мутациями дефицит этого белка делает мышечные клетки и волокна крайне нестабильными и восприимчивыми к повреждениям. Хотя точные механизмы развития этой патологии все еще требуют дополнительных исследований, уже известно, что большую роль в потере рабочей активности мышечных клеток играет нарушение работы митохондрий. Ранее авторы статьи выяснили, что при болезни Дюшенна в скелетных мышцах появляются серьезные нарушения: митохондрии, производящие энергию и регулирующие транспорт ионов кальция в клетке, начинают работать гораздо менее эффективно. В то же время отмечается, что благодаря митохондриям, клетки сердца, напротив, способны на ранних этапах заболевания сдерживать развитие неблагоприятного сценария. В своей новой работе биологи из республики Марий Эл совместно с коллегами из подмосковного наукограда Пущино изучили влияние терапевтического агента дефлазакорта на работу митохондрий скелетных мышц мышей с точечной мутацией в гене дистрофина, а также здоровых животных, использованных в качестве контроля.
«Сегодня одним из путей коррекции мышечной дистрофии Дюшенна является применение глюкокортикоидов, в частности преднизона и его оксазолинового производного дефлазакорта. Ряд других подходов, связанных с подавлением миостатина и применением микродистрофиновой терапии, находятся на стадии испытаний и показывают многообещающие результаты. Однако сейчас дефлазакорт – единственный препарат, официально одобренный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов в США (FDA) для лечения мышечной дистрофии Дюшенна. Терапия на основе этого глюкокортикоида, обладающего противовоспалительным действием, продлевает двигательную активность пациентов на 2–5 лет, а также улучшает мышечную силу и сердечно-легочную функцию. Кроме того, этот препарат обладает гораздо более «мягкими» побочными эффектами, по сравнению с предшественниками», — рассказывает Михаил Дубинин, руководитель проекта по гранту РНФ, кандидат биологических наук, доцент кафедры биохимии, клеточной биологии и микробиологии Марийского государственного университета (Йошкар-Ола).

В ходе исследования ученые в течение месяца вводили дефлазакорт модельным дистрофин-дефицитным мышам, а также здоровым животным, для того чтобы выявить как положительные, так и отрицательные эффекты такой терапии на работу скелетной мускулатуры и активность митохондрий мышц. Оказалось, что лечение мышей, страдающих мышечной дистрофией, с применением дефлазакорта сопровождается улучшением функциональной активности митохондрий скелетных мышц – они начинают гораздо более активно выполнять свои основные функции, а именно синтезировать АТФ – основную энергетическую молекулу всех живых клеток. Кроме того, они более интенсивно регулируют транспорт ионов кальция, что необходимо для поддерживания сократительной функции скелетной мускулатуры. В конце эксперимента ученые также оценили физическую силу и выносливость мышей с помощью теста на струне – оказалось, что дистрофин-дефицитные мыши, получавшие дефлазакорт, были способны гораздо дольше висеть на струне, держась за нее передними лапами.
«Стоит отметить, что мы обнаружили и побочные эффекты такой терапии. Действительно, митохондрии скелетных мышц мышей, получавших инъекции этого глюкокортикоида, способны гораздо эффективнее поглощать избыток ионов кальция, возникающий в мышечной клетке при дистрофии Дюшенна. Однако в этом случае способность митохондрий удерживать накопленные ионы кальция уменьшалась. Важно отметить, что такой эффект наблюдался и у здоровых животных, получавших дефлазакорт, — говорит Михаил Дубинин. — Поэтому мы полагаем, что со временем такое действие этого глюкокортикоида может приводить к снижению эффективности терапии. Необходима грамотная и своевременная консультация специалистов, а также соблюдение необходимой дозировки при назначении этого препарата».
Исследование проводили сотрудники Марийского государственного университета и Института теоретической и экспериментальной биофизики (ИТЭБ) РАН.
Мышечная дистрофия Дюшенна | ООО «Геномед»
Этот тип мышечной дистрофии относится к прогрессирующим генетическим заболеваниям и характеризируется патогенными поражениями, некрозом мышечных волокон, замещением их жировой и соединительной тканью.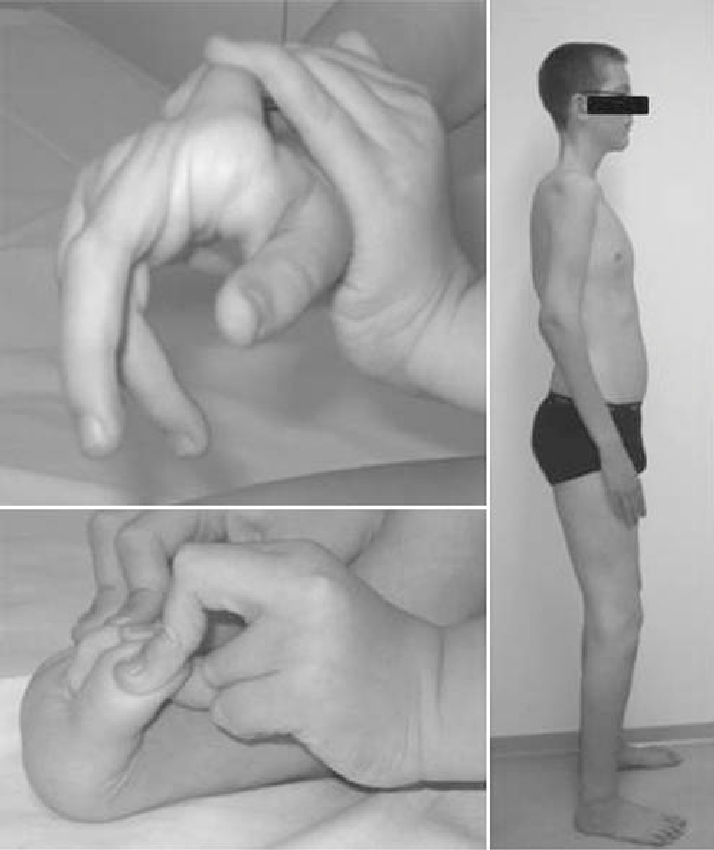
Симптоматика мышечной дистрофии Дюшенна
До 2-3 лет ребенок может не отставать в развитии от сверстников, первые симптомы проявляются на 3-5 году жизни, когда возникает мышечная слабость и утомляемость после физических нагрузок.
Отчетливое прогрессирование симптомов особенно заметно к 7-8 годам: меняется походка, а ослабленные мышцы начинают ограничивать самостоятельное передвижение больных. К 14-15 годам двигательная активность полностью ограничивается.
Характерными клиническими признаками мышечной дистрофии Дюшенна являются:
- симметричность развития патологических нарушений восходящего типа: вначале поражаются проксимальные мышц ног, таза и бедер, затем атрофия распространяется в область спины, плечевого пояса, верхних конечностей;
- при пальпации отмечается болезненность, плотность мышечных волокон;
- ограничиваются пассивные движения в суставах из-за непроизвольных сокращений мышечной ткани, возникают сухожильные ретракции;
- миодистрофия Дюшенна сочетается с патологическими изменениями костей и суставов (деформируются стопы, позвоночник), дисфункциональными расстройствами нейроэндокринной и сердечно-сосудистой систем;
- умственная отсталость проявляется в 30% случаев.
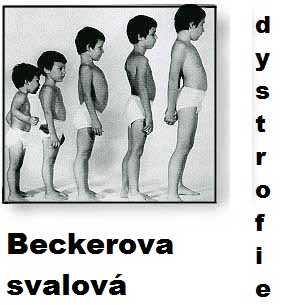
Этиология мышечной дистрофии Дюшенна
Возникновение и развитие патологии провоцирует генетическое поражение Х-хромосомы: нарушается синтез белка дистрофина, являющегося основой мышечных волокон и обеспечивающего их сокращение, пребывание в неактивном состоянии. При мышечной дистрофии этот белок не вырабатывается или продуцируется дефективным, что приводит к перерождению мышц и прогрессирующему ограничению двигательной активности.
Миодистрофия Дюшенна наследуется по рецессивному, сцепленному с Х-хромосомой типу. При этом девочки являются носителями патологии, а развивается заболевание только у лиц мужского пола (так как рецессивный аллель у них не подавляется доминантной парной генетически нормальной Х-хромосомой).
Диагностика мышечной дистрофии
- Аппаратные исследования: МРТ мышечной ткани применяется для определения степени поражений, ЭНМГ (электронейромиография) позволяет оценить функциональные характеристики периферической НС и мышц.
- Лабораторная диагностика: проводится биохимический АК на КФК (уровень креатинфосфокиназы) – увеличенная до 50 раз активность КФК при миодистрофических поражениях свидетельствует о прогрессировании заболевания.
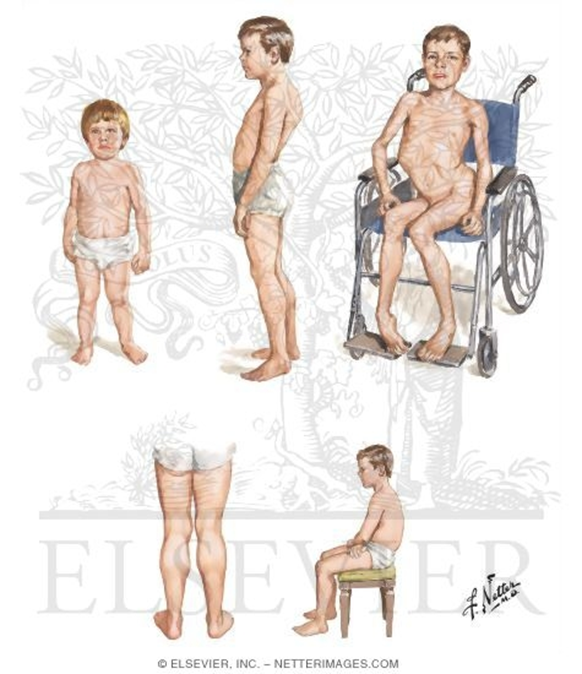
Но наиболее показательными и информативными являются генетические молекулярные исследования (панель «Нервно-мышечные заболевания», микроматричный анализ – ХМА), которые точно определяют нозологическую форму мышечной дистрофии.
Кроме того, около трети диагностируемых случаев миодистрофии Дюшенна имеют спорадический характер. Возникновение 6-7% спонтанных мутаций обусловлено органным гонадным мозаицизмом – наличием у матери генетически здоровых и мутантных первичных половых клеток.
Поэтому пренатальная молекулярная диагностика позволяет идентифицировать, какой именно структурный вариант гена (аллель) получил ребенок. Даже если установлен мужской пол эмбриона, вероятность передачи от матери мутантной популяции гамет не оценивается в 100%, как и генетический риск наследования идентичной мутации братьями (сестрами) больного со спорадической миодистрофией.
Лечение мышечной дистрофии Дюшенна
- Медикаментозная терапия: прием ингибиторов АПФ улучшает состояние мышц, аталурен восстанавливает продуцирование дистрофина, гормональные препараты замедляют прогрессирование симптомов заболевания.
- Наиболее перспективной на сегодняшний день является генная терапия (экзон-скиппинг), которая дает возможность проводить коррекцию мутации, провоцирующей развитие мышечной дистрофии, с помощью добавления донорского экзогенного участка молекулы ДНК.

Мышечная дистрофия Дюшенна: симптомы, диагностика и лечение
Что такое мышечная дистрофия Дюшенна?
Мышечные дистрофии — это группа заболеваний, которые со временем делают мышцы слабее и менее гибкими. Мышечная дистрофия Дюшенна (МДД) является наиболее распространенным типом. Это вызвано недостатками гена, который контролирует поддержание здоровья мышц в организме.
Заболевание почти всегда поражает мальчиков, и симптомы обычно проявляются в раннем детстве. Детям с МДД трудно вставать, ходить и подниматься по лестнице.Многие в конечном итоге нуждаются в инвалидных колясках для передвижения. У них также могут быть проблемы с сердцем и легкими.
Хотя лекарства и не существует, перспективы для людей с МДД лучше, чем когда-либо. Много лет назад дети с этим заболеванием обычно не доживали до подросткового возраста. Сегодня они доживают до 30, а иногда и от 40 до 50. Существуют методы лечения, которые могут облегчить симптомы, и исследователи также ищут новые.
Много лет назад дети с этим заболеванием обычно не доживали до подросткового возраста. Сегодня они доживают до 30, а иногда и от 40 до 50. Существуют методы лечения, которые могут облегчить симптомы, и исследователи также ищут новые.
Причины
МДД вызван проблемой в одном из ваших генов.Гены содержат информацию, необходимую вашему организму для производства белков, которые выполняют множество различных функций организма.
Продолжение
Если у вас МДД, ген, вырабатывающий белок, называемый дистрофином, нарушен. Этот белок обычно сохраняет мышцы сильными и защищает их от травм.
Заболевание чаще встречается у мальчиков из-за того, как родители передают гены МДД своим детям. Это то, что ученые называют заболеванием, связанным с полом, потому что оно связано с группами генов, называемыми хромосомами, которые определяют, мальчик или девочка.
Это редко, но иногда люди, в семейном анамнезе которых не было МДД, заболевают, когда их гены дефектны сами по себе.
Симптомы
Если у вашего ребенка МДД, вы, вероятно, заметите первые признаки до того, как ему исполнится 6 лет. Мышцы ног обычно страдают одними из первых, поэтому они, вероятно, начнут ходить намного позже, чем другие дети их возраста. Когда они научатся ходить, они могут часто падать и испытывать проблемы с подъемом по лестнице или поднятием с пола.Через несколько лет они также могут начать ходить вперевалку или ходить на цыпочках.
Продолжение
МДД также может повредить сердце, легкие и другие части тела. По мере взросления у вашего ребенка могут появиться другие симптомы, в том числе:
- Искривленный позвоночник, также называемый сколиозом
- Укороченные, напряженные мышцы ног, называемые контрактурами
- Головные боли
- Проблемы с обучением и памятью
- Кратковременность дыхание
- Сонливость
- Проблемы с концентрацией внимания
Проблемы с мышцами иногда могут вызывать судороги, но в целом МДД не вызывает болезненных ощущений.Ваш ребенок по-прежнему будет контролировать свой мочевой пузырь и кишечник. Хотя у некоторых детей с этим расстройством есть проблемы с обучением и поведением, МДД не влияет на интеллект вашего ребенка.
Получение диагноза
Вы должны сообщить врачу вашего ребенка о симптомах, которые вы заметили. Они захотят узнать историю болезни вашего ребенка, а затем задать вопросы о его симптомах, например:
- Сколько лет было вашему ребенку, когда он начал ходить?
- Насколько хорошо они умеют бегать, подниматься по лестнице или вставать с пола?
- Как давно вы замечаете эти проблемы?
- Есть ли у кого-нибудь в вашей семье мышечная дистрофия? Если да, то какие?
- Есть ли у них проблемы с дыханием?
- Насколько хорошо они обращают внимание или запоминают вещи?
Продолжение
Врач проведет физический осмотр вашего ребенка, и он может сделать некоторые тесты, чтобы исключить другие состояния, которые могут вызвать мышечную слабость.
Если врач подозревает МДД, он проведет другие анализы, в том числе:
- Анализы крови. Врач возьмет образец крови вашего ребенка и проверит его на креатинкиназу, фермент, который выделяется вашими мышцами при их повреждении. Высокий уровень КК является признаком того, что у вашего ребенка может быть МДД.
- Генные тесты. Врачи также могут проанализировать образец крови, чтобы найти изменение в гене дистрофина, которое вызывает МДД. Девочки в семье могут пройти тест, чтобы узнать, несут ли они этот ген.
- Биопсия мышц. С помощью иглы врач удаляет крошечный кусок мышцы вашего ребенка. Они будут смотреть на него под микроскопом, чтобы проверить низкий уровень дистрофина, белка, который отсутствует у людей с МДД.
Вопросы для врача
Если у вашего ребенка МДД, вам нужно получить как можно больше информации о его состоянии. Подумайте о том, чтобы спросить:
- Что это значит для моего ребенка?
- Нужно ли им обращаться к другим врачам?
- Какие существуют виды лечения?
- Как они будут себя чувствовать?
- Как я могу помочь им быть активными?
- Какую диету им следует придерживаться?
Лечение
Лекарства от МДД не существует, но существуют лекарства и другие методы лечения, которые могут облегчить симптомы вашего ребенка, защитить его мышцы и сохранить здоровье сердца и легких.
Этеплирсен (Exondys 51) был одобрен для лечения людей со специфической мутацией гена, приводящей к МДД. Это лекарство для инъекций, которое помогает. Наиболее частыми побочными эффектами являются нарушение равновесия и рвота. Хотя препарат увеличивает выработку дистрофина, что предсказывает улучшение мышечной функции, это еще не было показано.
Пероральный кортикостероид дефлазакорт (Эмфлаза) был одобрен в 2017 году для лечения МДД, став первым одобренным FDA кортикостероидом для лечения этого состояния.Было обнаружено, что дефлазакорт помогает пациентам сохранять мышечную силу, а также помогает им сохранять способность ходить. Общие побочные эффекты включают отечность, повышенный аппетит и увеличение веса.
Стероиды, такие как преднизон, замедляют мышечное повреждение. Дети, принимающие это лекарство, могут ходить на 2-5 лет дольше, чем без него. Эти препараты также могут улучшить работу сердца и легких вашего ребенка.
Продолжение
Поскольку МДД может вызвать проблемы с сердцем, важно, чтобы ваш ребенок посещал кардиолога, называемого кардиологом, для осмотров один раз в 2 года до 10 лет и один раз в год после этого.Девочки и женщины, несущие этот ген, также имеют более высокий риск сердечных заболеваний. В подростковом или раннем взрослом возрасте им следует обратиться к кардиологу, чтобы проверить наличие каких-либо проблем.
Для небольшого числа пациентов с МДД с пропущенным экзоном 45 генной мутации был одобрен инъекционный казимерсен (Amondys 45). Это первое целевое лечение этого типа мутации, которое, как было показано, помогает увеличить выработку дистофина.
Продолжение
У небольшого числа детей с МДД также может быть генная мутация, поддающаяся пропаданию экзона 53.Лекарство голодирсен (Vyondys 53) было одобрено для увеличения количества дистофина в мышечных волокнах.
Некоторые лекарства от артериального давления могут помочь защитить сердечные мышцы от повреждения.
Продолжение
Детям с МДД может потребоваться операция для исправления укороченных мышц, выпрямления позвоночника или лечения проблем с сердцем или легкими.
Ученые продолжают искать новые способы лечения МДД в рамках клинических испытаний. Эти испытания проверяют новые лекарства, чтобы убедиться, что они безопасны и работают.Часто они позволяют людям попробовать новое лекарство, доступное далеко не каждому. Ваш врач может сказать вам, подходит ли одно из этих испытаний для вашего ребенка.
Забота о ребенке
Удивительно узнать, что у вашего ребенка МДД. Помните, что болезнь не означает, что они не могут ходить в школу, заниматься спортом и веселиться с друзьями. Если вы будете придерживаться их плана лечения и знаете, что работает для вашего ребенка, вы можете помочь ему вести активный образ жизни.
- Стойте и ходите как можно больше. Если вы будете стоять в вертикальном положении, кости вашего ребенка будут крепкими, а позвоночник будет прямым. Подтяжки или ходунки могут помочь им встать и передвигаться.
- Правильно питайтесь. Специальной диеты для детей с МДД не существует, но здоровая пища может предотвратить проблемы с весом или помочь при запорах. Проконсультируйтесь с диетологом, чтобы убедиться, что ваш ребенок ест правильный баланс питательных веществ и калорий каждый день. Возможно, вам потребуется обратиться к специалисту, если у вашего ребенка проблемы с глотанием.
- Оставайтесь активными. Упражнения и растяжка помогут сохранить гибкость мышц и суставов вашего ребенка и улучшить его самочувствие. Физиотерапевт может научить их, как безопасно выполнять упражнения, не перегружая себя работой.
- Обратитесь за поддержкой. Другие семьи, живущие с МДД, могут быть отличным источником совета и понимания жизни с этим заболеванием. Найдите местную группу поддержки или изучите онлайн-форумы. Также вам может быть полезно поговорить о своих чувствах с психологом или консультантом.
Чего ожидать
Когда ваш ребенок станет старше, его мышцы станут слабее, и он, скорее всего, не сможет ходить.Многим мальчикам с МДД к 12 годам понадобится инвалидная коляска, которая поможет им передвигаться. Хотя некоторые дети доживают до подросткового возраста, перспективы этого состояния намного лучше, чем раньше. Сегодня молодые люди с МДД могут поступать в колледж, делать карьеру, жениться и создавать семьи.
Ученые также исследуют новые способы лечения генов, вызывающих МДД. Эти методы лечения могут вскоре улучшить перспективы для людей с МДД. В 2019 году FDA одобрило инъекцию голодирсена (Vyondys 53,) в качестве первого препарата для лечения МДД у пациентов с подтвержденной мутацией, поддающейся пропуску экзона 53, а в 2014 году официальные лица в Европе одобрили аталурен (Трансларна) в качестве первого препарата для лечения генетического причина МДД.Несколько других генных методов лечения могут быть скоро готовы к продаже в США
Получение поддержки
Чтобы узнать больше о мышечной дистрофии Дюшенна или найти группу поддержки в вашем районе, посетите: Cure Duchenne, Ассоциацию мышечной дистрофии или Родительский проект Мышечная дистрофия.
Заболевания — МДД — Диагноз
Диагностика
При диагностике любой формы мышечной дистрофии врач обычно начинает с изучения истории болезни пациента и его семьи, а также проведения физического обследования.Врачи могут обнаружить псевдогипертрофию, отклонение поясничного отдела позвоночника, аномалии походки и несколько степеней снижения мышечных рефлексов.
Из этих наблюдений можно многому научиться, включая характер слабости. Анамнез пациента и его физическое состояние имеют большое значение для постановки диагноза, даже до того, как будут выполнены какие-либо сложные диагностические тесты.
Кардиомиопатия у пациентов с МДД также может быть связана с нарушениями проводимости. Врач может заметить характерные изменения на электрокардиограмме.Кроме того, с помощью эхокардиографии можно обнаружить структурные изменения сердца, такие как порок клапанов сердца (особенно поражающий митральный клапан, когда он возникает). Поэтому необходимы электрокардиограмма, неинвазивная визуализация с эхокардиографией или МРТ сердца, а также консультация кардиолога.
Уровни СК и других ферментов
В начале диагностического процесса врачи часто назначают анализ крови под названием CK level . CK означает креатинкиназу , фермент, который выделяется из поврежденных мышц.Когда в образце крови обнаруживается повышенный уровень КФК, это обычно означает, что мышца разрушается в результате какого-либо аномального процесса, такого как мышечная дистрофия или воспаление. Очень высокий уровень CK предполагает, что сами мышцы (а не нервы, которые их контролируют) являются вероятной причиной слабости, хотя это не указывает на то, какой именно тип мышечного расстройства может иметь место. Высокий уровень КФК может быть обнаружен до появления симптомов даже у новорожденных, страдающих МДД.
Пик уровня CK (в 10-20 раз превышающий верхнее предельное значение) к возрасту 2 лет, затем постепенно снижается со скоростью 25% в год, в конечном итоге возвращаясь к нормальному уровню, когда значительное количество мышечной ткани заменяется жиром и рубцовая / фиброзная ткань.
Генетическое тестирование
Генетическое тестирование включает анализ ДНК любых клеток (обычно используются клетки крови), чтобы увидеть, есть ли мутация в гене дистрофина, и если да, то где именно она возникает. Такое ДНК-тестирование на мутации дистрофина широко доступно в Соединенных Штатах. Ваш врач Центра медицинского обслуживания MDA или консультант по генетическим вопросам может предоставить вам дополнительную информацию о вариантах тестирования. А чтобы узнать больше о точном генетическом диагнозе, см. «Джинн из бутылки: генетическое тестирование в 21 веке».
Обычно генетическая диагностика показана пациентам с повышенными уровнями КФК в сыворотке и клиническими проявлениями дистрофинопатии. Диагноз подтверждается, если выявлена мутация гена DMD . Генетический анализ в первую очередь направлен на обнаружение крупных делеционных / дупликационных мутаций (в 70–80% случаев присутствуют такие мутации). Если первоначальный генетический анализ отрицательный, следующим следует анализ небольших генных мутаций и генных микроделеций / дупликаций.
Женщины-родственники мужчин и мальчиков с МДД могут пройти ДНК-тестирование, чтобы определить, являются ли они носителями этой болезни.Женщины, являющиеся носителями МДД, могут передать болезнь своим сыновьям, а их статус носителя — своим дочерям. В меньшинстве случаев девочки и женщины, являющиеся носителями МДД, могут сами проявлять симптомы МДД, такие как мышечная слабость и проблемы с сердцем. Эти симптомы могут не появиться до взрослого возраста (см. Причины / Наследование).
Несколько экспериментальных препаратов, которые в настоящее время разрабатываются для лечения МДД, требуют знания точной генетической мутации человека, поэтому генетическое тестирование стало важным не только для диагностики, но, возможно, и для будущего лечения.
Биопсия мышцы
Для получения дополнительной информации врач может назначить биопсию мышцы , хирургическое удаление небольшого образца мышцы у пациента. Изучая этот образец, врачи могут многое рассказать о том, что на самом деле происходит внутри мышц. Однако в современную эпоху биопсия мышц требуется редко, потому что почти всем пациентам ставится диагноз генетического тестирования.
Современные методы могут использовать биопсию, чтобы отличать мышечные дистрофии от воспалительных и других заболеваний, а также различать различные формы мышечной дистрофии.Например, количество функционального белка-дистрофина, обнаруженного в образце мышечной биопсии, проливает свет на то, вероятно ли течение заболевания — МДД без наличия дистрофина или более легкая мышечная дистрофия Беккера (МПД) с присутствием некоторого частично функционального дистрофина.
Гистологические (тканевые) признаки миопатии могут наблюдаться с рождения у детей мужского пола с МДД. Хотя эндомиокардиальная биопсия (внутренний клеточный слой сердца) обычно не проводится, она показывает переменное распределение дистрофина в кардиомиоцитах (клетках сердечной мышцы).
По сравнению с МДД, МПК обычно начинается в более позднем возрасте (от 5 до 60 лет). Клиническое поражение, как правило, более легкое, с некоторой степенью сохранения силы. 1 Пациенты с МПК остаются амбулаторными как минимум до 16 лет, а в некоторых случаях и до взрослого возраста. Контрактуры и когнитивные расстройства менее распространены и тяжелы у пациентов с МПК по сравнению с пациентами с МДД. У пациентов с МПК уровни КК обычно повышены в пять и более раз. Вовлечение сердца в МПК часто является преобладающим.Пациенты с МПК обычно доживают до 30 лет.
Если подозрение на МДД остается высоким, несмотря на отрицательный генетический анализ, проводится определение дистрофина методом вестерн-блоттинга или окрашивания селективными антителами в ткани, полученной при биопсии мышц. Вестерн-блоттинг полезен для прогнозирования тяжести заболевания, поскольку количество дистрофина, присутствующего в анализе, зависит от клинической картины. Менее 5% от нормального количества дистрофина связано с МДД, уровень от 5% до 20% от нормы относится к промежуточному заболеванию, и более 20% от нормального уровня связан с МПК. 2,3
Список литературы
- Брэдли, У. Г., Джонс, М. З., Муссини, Дж. -М и Фосетт, П. Р. У. Мышечная дистрофия по типу Беккера. Мышечный нерв (1978). DOI: 10.1002 / mus.880010204
- Hoffman, E. P. et al. Характеристика дистрофина в образцах мышечной биопсии от пациентов с мышечной дистрофией Дюшенна или Беккера. N. Engl. J. Med. (1988). DOI: 10.1056 / NEJM198805263182104
- Hoffman, E. P. et al. Улучшенная диагностика мышечной дистрофии Беккера с помощью тестирования на дистрофин. Неврология (2012). DOI: 10.1212 / wnl.39.8.1011
Медикаментозное лечение мышечной ткани Дюшенна имеющиеся данные и перспективы
Acta Myol. 2012 May; 31 (1): 4–8.
Отделение нейропедиатрии и мышечных заболеваний, Университетский медицинский центр Фрайбурга, Германия
Адрес для корреспонденции: Янбернд Киршнер, Отделение нейропедиатрии и мышечных заболеваний, Университетский медицинский центр Фрайбурга, Mathildenstrasse 1, 79106 Freiburg, Германия.Электронное письмо: [email protected] Авторское право Журнал и отдельные статьи, содержащиеся в нем, защищены авторским правом Академии Гаэтано Конте, Неаполь, Италия Это статья в открытом доступе, распространяемая в соответствии с условиями некоммерческой лицензии Creative Commons Attribution Non-Derivatives License, которая разрешает некоммерческое использование, распространение и воспроизведение на любом цифровом носителе при условии, что оригинальная работа должным образом процитирована и не будет изменена в каких-либо способ. Подробнее см. Http: // creativecommons.org / licenses / by-nc-nd / 3.0 / Эта статья цитируется в других статьях PMC.Abstract
Мышечная дистрофия Дюшенна (МДД) — это заболевание, связанное с Х-хромосомой, которое поражает 1 из 3 600-6 000 новорожденных мальчиков. Это проявляется в отсутствии белка дистрофина в мышечных волокнах, который вызывает прогрессирующее повреждение, ведущее к смерти на третьем десятилетии жизни. Единственными лекарствами, которые до сих пор доказали свою эффективность в замедлении прогрессирования этого заболевания, являются кортикостероиды, которые, как было показано в рандомизированных контролируемых исследованиях, увеличивают мышечную силу; Долгосрочные исследования показали, что они увеличивают время ходьбы и замедляют прогрессирование респираторной дисфункции, дилатационной кардиомиопатии и сколиоза.Несколько потенциальных лекарств сейчас исследуются. Генетическая терапия, включающая введение гена дистрофина через вектор, доказала свою эффективность на животных, но не на людях. В настоящее время проходит клиническое исследование Аталурена, молекулы, которая связывается с рибосомами и может допускать вставку аминокислоты в кодон преждевременной терминации и пропуск экзона, который связывается с РНК и исключает определенные сайты сплайсинга РНК, производя дистрофин меньшего размера. но функциональный. Есть также исследования, пытающиеся модулировать другие мышечные белки, такие как миостатин и атрофин, для уменьшения симптомов.В этой статье не рассматривается лечение кардиомиопатии у пациентов с МДД.
Ключевые слова: мышечная дистрофия Дюшенна, медикаментозное лечение, клиника. испытания
Введение
Мышечная дистрофия Дюшенна (МДД) — это заболевание, связанное с Х-хромосомой, поражающее 1 из 3 600-6 000 новорожденных мальчиков. Он характеризуется слабостью проксимальных мышц, выражающейся в положительном знаке Гауэрса при вставании, аномальной походкой, гипертрофией икроножных мышц и повышенным уровнем креатинкиназы.Большинству пациентов диагноз ставится в возрасте 5 лет, когда симптомы становятся более очевидными. Заболевание имеет прогрессирующее течение мышечной слабости, поражающей также сердечные мышцы и дыхательную систему. Больные мальчики обычно прекращают ходить в возрасте 13 лет и, если их не лечить, умирают до 20 лет от сердечных заболеваний или респираторных инфекций (1, 2).
DMD вызывается мутациями в гене дистрофина, что приводит к серьезному снижению или полному отсутствию белка дистрофина, который обнаруживается в сарколемме мышечных волокон и состоит из четырех частей: C-концевого домена, который присоединяется к другим белкам в мышечных волокнах. мембрана, называемая комплексом дистрогликана, стержневым доменом, богатым цистеином доменом и N-концевым доменом, который связывается с актином (3).Таким образом, С-концевой домен взаимодействует как мост между сарколеммой и внеклеточным матриксом и связывается с другими мембранными белками через дистрофин-гликопротеиновый комплекс. Предполагается, что дистрофин необходим для преобразования силы сократительного аппарата во внеклеточный матрикс и защищает мышечные волокна от любого повреждения, вызванного сокращением мышц, которое может привести к некрозу (3, 4). Отсутствие дистрофина приводит к механическому повреждению сарколеммы, потере гомеостаза кальция и прогрессирующей дегенерации мышечных волокон.
Ген дистрофина — самый крупный ген, обнаруженный у человека, и на его долю приходится примерно 0,1% всего генома человека. Он расположен на коротком плече Х-хромосомы и состоит из 79 экзонов и 7 промоторных областей (4). Сообщаемая частота различных мутаций, приводящих к МДД, широко варьируется. По данным базы данных Лейдена (www.dmd.nl), дупликации одного или нескольких экзонов соответствуют 7% мутаций, точечные мутации составляют 20%, а делеции наблюдаются у 72% пациентов (4, 5).Большинство делеций происходит между экзонами 44 и 55, соответствующими стержневому домену дистрофина (6). Если эти мутации изменяют рамку считывания дистрофина (вне рамки мутации), образование белка прекращается, дистрофин не вырабатывается и у пациента развивается МДД. Если мутация находится «в рамке», дистрофин меньше по размеру, но все еще функционирует, и в этом случае пациенту ставится диагноз мышечной дистрофии Беккера (7).
Хотя молекулярное происхождение МДД известно уже несколько лет, до сих пор не существует лечебного лечения этого заболевания.На сегодняшний день единственным эффективным средством замедления прогрессирования заболевания являются кортикостероиды. Они изменили естественное течение МДД. Однако их точный механизм действия полностью не изучен (2).
Лекарственные препараты
Кортикостероиды: преднизон и дефлазакорт
Глюкокортикоиды, точнее преднизон и дефлазакорт, являются основными лекарственными средствами для лечения МДД. Они используются уже более двух десятилетий, и теперь их преимущества хорошо известны.Это единственное лекарство, которое увеличивает мышечную силу. Ранние исследования доказали, что их использование продлевает ходьбу и улучшает их функциональность в повседневной деятельности. Долгосрочные исследования показали, что они также уменьшают потребность в хирургии сколиоза, улучшают функцию легких и помогают поддерживать сердечную функцию (8, 9).
В 2005 г. Американская академия неврологии, а в 2008 г. — Кокрановский обзор, оценили все рандомизированные контролируемые испытания по применению кортикостероидов и пришли к выводу, что преднизон вводят в дозах 0.75 мг / кг / день показали увеличение мышечной силы и улучшение результатов в стандартизированных функциональных тестах в краткосрочной перспективе (10, 11). Увеличение мышечной силы происходит в течение первых шести месяцев лечения, после чего следует период стабилизации в течение двух лет с последующим снижением, которое происходит медленнее, чем у нелеченных пациентов (12).
В пяти недавно опубликованных долгосрочных контролируемых нерандомизированных исследованиях (продолжительностью более 3 лет) с преднизоном или дефлазакортом сообщалось, что после приема одного из этих препаратов пациенты могут ходить на 2-5 лет дольше, чем те, кто не получает кортикостероиды, необходимость стабилизации позвоночника. количество операций сократилось, а потребность в неинвазивной вентиляции легких отпала (8).Два из этих исследований оценивали сердечную функцию и продемонстрировали, что фракция выброса левого желудочка у пролеченных пациентов была значительно лучше сохранена по сравнению с нелеченными пациентами (8). Другое исследование показало, что 93% пациентов, получавших преднизолон, не имели желудочковой дисфункции в возрасте 12 лет по сравнению с 53% пациентов, не получавших лечения (13). Исследования также показывают увеличение продолжительности жизни (14). Игл упоминает, что в 1960 году средняя продолжительность жизни пациентов с МДД составляла 14 лет.4 года, которые к 1990 году выросли до 19,3 года с использованием кортикостероидов, антибиотикотерапии и интенсивной терапии. В настоящее время он увеличился до 24,5 лет, вероятно, из-за использования неинвазивной вентиляции (8, 14).
Таким образом, есть существенные доказательства, позволяющие рекомендовать использование кортикостероидов всем пациентам с МДД с целью сохранения времени ходьбы как можно дольше и уменьшения легочных, сердечных и ортопедических осложнений (2). Естественно, следует учитывать и побочные эффекты лечения кортикостероидами.Наиболее частым побочным эффектом при длительном лечении является уменьшение роста пациента. Увеличение веса является второй по частоте, но основной причиной прекращения лечения (8). Увеличение веса у пациентов с МДД, получающих стероидные препараты, является многофакторной проблемой. Это не только побочный эффект кортикостероидов — это также результат их ограниченной подвижности, поскольку увеличение веса обычно более выражено у неамбулаторных пациентов. Дефлазакорт вызывал меньшую прибавку в весе, но больше катаракт, чем преднизон.Частота переломов позвонков у пролеченных пациентов варьировала на 5-32%. Около 80% этих переломов были обнаружены при рутинных рентгенологических исследованиях сколиоза и не сопровождались клиническими симптомами (8, 15). К другим побочным эффектам относятся кушингоидная фация, угри, гирсутизм, артериальная гипертензия, расстройство поведения, задержка полового созревания, переломы позвонков, иммуносупрессия и желудочно-кишечные проблемы. Хотя эти побочные эффекты менее часты при использовании в указанных дозах, тем не менее, они являются условиями, которые необходимо контролировать.
Указанная доза преднизона составляет 0,75 мг / кг в сутки. Дозы менее 0,3 мг / кг менее эффективны, а ежедневное введение кажется более эффективным, чем через день (10).
Прерывистые режимы постулируются как имеющие лучший профиль безопасности с точки зрения побочных эффектов, но данные единственного рандомизированного контролируемого исследования прерывистого преднизона были недоступны (что позволило бы провести статистический анализ и сравнение с другими режимами). В одном открытом когортном исследовании с периодическим применением дефлазакорта сообщалось о продлении передвижения (12).Следовательно, необходимы рандомизированные контролируемые исследования для определения оптимального графика лечения в долгосрочной перспективе.
В настоящее время рекомендуется начинать лечение, когда пациент находится в «фазе плато». Обычно это происходит в возрасте от 4 до 6 лет, когда мальчик перестает двигать вперед. В настоящее время многие врачи продолжают лечение даже после того, как пациент с МДД потерял способность ходить, с целью сохранения функции верхних конечностей, снижения скорости прогрессирования сколиоза и замедления нарушения дыхательной и сердечной функции ( 2).По-прежнему необходимы дальнейшие исследования, чтобы определить, продолжают ли неамбулаторные пациенты получать пользу от этого лечения.
Дефлазакорт представляет собой оксазолиновое производное преднизона и в дозе 0,9 мг / кг / день так же эффективен, как и преднизон, при лечении МДД. Выбор зависит от местной доступности дефлазакорта и преднизона, его стоимости, состава и предпочтений пациента. Одно небольшое рандомизированное исследование предполагает более высокую частоту и выраженность увеличения веса при приеме преднизона, чем при применении дефлазакорта (13).Таким образом, некоторые пациенты предпочитают дефлазакорт. Однако риск развития катаракты повышен по сравнению с преднизоном: в нерандомизированных исследованиях 10-30% катаракт наблюдались в среднем через 3,2 года лечения дефлазакортом. Таким образом, эти пациенты должны ежегодно наблюдаться офтальмологом (2, 12).
Другие иммунодепрессанты
Было высказано предположение, что польза от лечения кортикостероидами может быть связана с иммунодепрессивным эффектом.Таким образом, были проведены дальнейшие исследования с другими иммунодепрессантами. Однако рандомизированное контролируемое исследование 99 мальчиков с МДД, получавших только азатиприн или в комбинации с преднизоном, показало, что азатиоприн не имеет положительного эффекта (16). Плацебоконтролируемое двойное слепое исследование 146 амбулаторных пациентов с МДД, получавших только циклоспорин-А или плацебо в сочетании с преднизоном, не выявило разницы в силе мышц и функциональных возможностях между группами лечения (17).
Генетическая терапия
Недавно были проведены исследования возможностей генетической терапии, посредством которой встраивается ген дистрофина. Однако на этом пути встретилось несколько препятствий. Размер гена дистрофина затрудняет работу с ним в генной терапии. Таким образом, были разработаны гены меньшего размера, микро- или мини-дистрофин, которые можно вставить в вектор. Наиболее подходящим вектором, обнаруженным на данный момент, является вирус, связанный с аденовирусом, непатогенным парвовирусом, но было показано, что он вызывает иммунологический ответ.Чтобы оценить ответ, были созданы mdx мышей dys- / dys-, и есть свидетельства того, что при инъекции гена дистрофин частично экспрессируется, а мышечная сила повышается. Однако в предварительных исследованиях на людях через 90 дней после начала лечения экспрессия этого гена не наблюдалась. Результаты показывают, что клеточный иммунитет препятствует успеху этой терапии (18, 19).
Пропуск экзона
При мышечной дистрофии Беккера белок дистрофина меньше по размеру и частично функционирует, что приводит к менее тяжелому заболеванию.У пациентов с МДД, как упоминалось ранее, мутировавший ген проявляет делеции, дупликации и точечные мутации, которые нарушают рамку считывания генетической информации. В настоящее время исследователи стремятся ввести молекулу, способную вмешиваться в сигналы сплайсинга РНК, чтобы пропустить дополнительный соседний экзон, тем самым восстанавливая рамку считывания и обеспечивая экспрессию белка меньшего размера, но частично функционального, как у пациентов, у которых есть Мышечная дистрофия Беккера. Эта синтетическая модифицированная молекула РНК называется антисмысловым олигонуклеотидом (АО) и способна связываться со специфическими участками пре-РНК, маскируя и исключая этот экзон из сплайсинга (5).Эта терапия будет индивидуальной для каждой мутации. Текущие исследования пропуска экзона направлены на пропуск экзона 51, «пропуск» которого применим к самой большой группе пациентов, включающей 13% всех пациентов с МДД, за которыми следуют мутации в экзоне 45. В настоящее время изучаются два АО:
2′-O-метилфосфориоаты (2OMP): исследования первоначально проводились на мышах mdx с 2OMP на экзоне 23. Они показали присутствие дистрофина во многих волокнах скелетных мышц, но не в сердце (20). .Затем было проведено клиническое исследование четырех пациентов с МДД с PRO051 / GSK2402964 (2OMP, нацеленным на экзон 51), и исследователи обнаружили, что 64-97% мышечных волокон экспрессируют белок дистрофин в количестве 17-35%. Побочных эффектов не наблюдалось (21). В настоящее время проводится многоцентровое исследование (22, 23).
Морфолиноолигомер фофородиамидата (PMO): исследования на мышах MDX показали присутствие дистрофина не только в мышечных волокнах, но и в сердце после введения высоких доз препарата.Длительное повторяющееся лечение даже продемонстрировало улучшение двигательной функции (24). На людях исследование экзона 51 у семи пациентов с МДД (AVI-4658) показало, что те, кто получал высокие дозы (0,9 мг), продуцировали дистрофин на 22-32% нормальных уровнях в 44-79% мышечных волокон. В этом исследовании не наблюдалось никаких признаков токсичности (25), в то время как предыдущее, проведенное на нечеловеческих приматах, показало дегенерацию канальцев в почках (26). Недостатком этого препарата является то, что эффект носит временный характер и ограничен временем, в течение которого АО остается в ткани.Кроме того, он производит уровни дистрофина ниже 30-60% от нормы, что, как предполагается, достаточно для компенсации мышечной дисфункции (27).
Преждевременное считывание стоп-кодона: аминогликозиды и аталурен
Эта форма лечения применяется только к мальчикам с мутациями, приводящими к преждевременным кодонам терминации, которые, по оценкам, встречаются примерно у 13-15% популяции МДД. Было показано, что считывание стоп-кодона способно подавлять кодоны терминации за счет неправильного считывания РНК, тем самым позволяя вставку альтернативных аминокислот в сайт мутировавшего кодона преждевременной терминации.В результате образуется полноразмерный белок-дистрофин с заменой только одной аминокислоты.
Аминогликозиды
В культурах клеток гентамицин взаимодействует с 40-й рибосомной субъединицей при транскрипции РНК, подавляя терминальные кодоны и вставляя на их место другую заменяющую ее аминокислоту. В исследованиях на мышах mdx и на людях гентамицин был способен вызывать экспрессию дистрофина в мышечных волокнах на 20% от нормального уровня (5). Однако исследования пациентов с МДД остаются противоречивыми.Фактически одно из них показало положительный эффект мышечной силы и повторную экспрессию дистрофина в мышцах (28), в то время как в другом исследовании с участием 12 пациентов с МДД, проведенном в течение шести месяцев, экспрессия дистрофина была обнаружена только в 6 из них. у 12 пациентов, и никаких клинических преимуществ не наблюдалось (29-30).
Аталурен (PTC124)
Аталурен — это пероральный препарат, который теоретически имеет тот же эффект, что и гентамицин, но который связан с 60-й рибосомной субъединицей.Его эффективность у мышей mdx аналогична гентамицину, вызывая экспрессию дистрофина в 20-25% мышечных волокон (5).
С этими результатами было проведено двойное слепое рандомизированное многоцентровое исследование с участием 174 пациентов. После 48 недель приема низких доз аталурена пациенты показали некоторое улучшение в тесте с 6-минутной ходьбой, но окончательные результаты не были опубликованы.
Миостатин
Миостатин — это белок, входящий в семейство трансформирующих факторов роста B, регулирующих размер мышц.Поскольку мышечная гипертрофия наблюдалась у мышей с нокаутом гена миостатина ( mys- / mys- ), блокировка миостатина может служить потенциальным средством лечения МДД. Было предложено несколько способов блокирования миостатина: фолластатин, блокатор рецепторов миостатина и деструктивный пропуск экзона для гена миостатина (5).
Антитело миостатина вводили мышам MDX, вызывая мышечную гипертрофию и большую силу. Только одно клиническое исследование было проведено на людях с антителом против миостатина (MYO-029) у взрослых пациентов с мышечной дистрофией, которое хорошо переносилось, но не увеличивало мышечную силу (32).
Утрофин
Утрофин разделяет 80% последовательности дистрофина и сверхэкспрессируется в мышцах пациентов с МДД. Было высказано предположение, что его повышенная регуляция может замедлить прогрессирование болезни. Инъекция херегулина, ларгинина или оксида азота мышам MDX увеличивает экспрессию утрофина в гистологических препаратах (33), а внутрибрюшинное введение белка TAT-утрофина увеличивает мышечную силу у крыс MDX. Было проведено исследование на здоровых людях с использованием препарата, разработанного Summit PLC (C110 / BM195), который оказался хорошо переносимым, хотя фармакокинетика препарата не позволяла продолжить лечение (5).
Добавка витамина D
Пациенты с МДД имеют повышенный риск развития патологических переломов длинных костей и позвоночника. Во-первых, минеральная плотность костной ткани снижается как следствие ограниченной подвижности. Во-вторых, риск остеопороза увеличивается из-за хорошо известного побочного эффекта длительного лечения кортикостероидами. В-третьих, они, как правило, имеют низкий уровень витамина D, вероятно, из-за меньшего воздействия солнечного света. Поэтому рекомендуются физические упражнения и достаточное количество кальция и витамина D в рационе.Добавки витамина D рекомендуется пациентам с доказанным дефицитом витамина D (сывороточный 25-гидроксивитамин D <50 мг / мл) (2, 35).
Нет данных о том, что бисфосфаты следует использовать с профилактической целью у детей, получающих стероидную терапию, однако введение этого препарата рекомендуется при возникновении патологических переломов (7, 35, 36).
Заключение
Хотя молекулярная основа мышечной дистрофии Дюшенна известна уже несколько десятилетий, до сих пор не существует лечения, которое излечивает эту болезнь.В настоящее время ведутся активные исследования по этой теме. До сих пор единственным лекарством, способным замедлить прогрессирование этого заболевания, были кортикостероиды.
Список литературы
1. Пойски Дж. Паттерны поведения при мышечной дистрофии Дюшенна: отчет на семинаре по поведению «Мышечная дистрофия» родительского проекта 8-9 декабря 2006 г., Филадельфия, США. Нервно-мышечное расстройство. 2007; 17: 986–994. [PubMed] [Google Scholar] 2. Бушби К., Финкель Р., Бирнкрант Д. Д. и др. Диагностика и лечение мышечной дистрофии Дюшенна, часть 1: диагностика и фармакологическое и психосоциальное управление.Lancet Neurol. 2010; 9: 77–93. [PubMed] [Google Scholar] 3. О’Брайен К.Ф., Кункель Л.М. Дистрофин и мышечная дистрофия: в прошлом, настоящее и будущее. Mol Genet Metab. 2001. 74: 75–88. [PubMed] [Google Scholar] 4. Мунтони Ф., Торелли С., Ферлини А. и др. Дистрофин и мутации: один ген, несколько белков, несколько фенотипов. Lancet Neurol. 2003; 2: 731–740. [PubMed] [Google Scholar] 5. Пичавант С., Аартсма-Рус А., Клеменс П.Р. и др. Текущее состояние фармацевтические и генетические терапевтические подходы к лечению МДД.2011. Mol Ther. 2011; 19: 830–840. [Бесплатная статья PMC] [PubMed] [Google Scholar] 6. Нельсон С.Ф., Кросби Р., Мичели М.К. и др. Новые генетические методы лечения для лечения мышечной дистрофии Дюшенна. Curr Opin Neurol. 2009; 22: 532–538. [Бесплатная статья PMC] [PubMed] [Google Scholar] 7. Манзур А.Ю., Кинали М., Мунтони Ф. Последние сведения об управлении Мышечная дистрофия Дюшенна. Arch Dis Child. 2008; 93: 986–990. [PubMed] [Google Scholar] 8. Moxley RT, Pandya S, Ciafoli E, et al. Изменения в естественной истории мышечной дистрофии Дюшенна при длительном приеме кортикостероидов лечение: значение для управления.J Child Neurol. 2010; 25: 1116–1129. [PubMed] [Google Scholar] 9. Houde S, Filiatrault M, Fournier A и др. Использование дефлазакорта при Дюшенне мышечная дистрофия: наблюдение через 8 лет. Pediatr Neurol. 2008. 38: 200–206. [PubMed] [Google Scholar] 10. Moxley RT, 3rd, Ashwal S, Pandya S, et al. Параметр практики: кортикостероид лечение дистрофии Дюшенна: отчет о качестве Подкомитет по стандартам Американской академии неврологии и Практический комитет Общества детской неврологии. Неврология.2005; 64: 13–20. [PubMed] [Google Scholar] 11. Анджелини Дж. Роль кортикостероидов при мышечной дистрофии: а Критическая оценка. Мышечный нерв. 2007. 36: 424–435. [PubMed] [Google Scholar] 12. Манзур А.Ю., Кунцер Т., Пайк М. и др. Глюкокортикоидные кортикостероиды при мышечной дистрофии Дюшенна. Кокрановская база данных Syst Ред. 2008 г .; 1: CD003725 – CD003725. [PubMed] [Google Scholar] 13. Маркхэм Л.В., Киннетт К., Вонг Б.Л. и др. Кортикостероидное лечение замедляет развитие желудочковой дисфункции в мышечной ткани Дюшенна. дистрофия.Нервно-мышечное расстройство. 2008. 18: 365–370. [PubMed] [Google Scholar] 14. Игл М., Бурк Дж., Баллок Р. и др. Управление мышечной массой Дюшенна дистрофия — аддитивный эффект хирургии позвоночника и домашнего ночная вентиляция для повышения выживаемости. Нервно-мышечное расстройство. 2007. 17: 470–475. [PubMed] [Google Scholar] 15. King WM, Ruttencutter R, Nagaraja HN и др. Ортопедические исходы длительного ежедневного лечения кортикостероидами при Дюшенне мышечная дистрофия. Неврология. 2007. 68: 1607–1613. [PubMed] [Google Scholar] 16.Griggs RC, Moxley RT, 3rd, Mendell JR, et al. Дистрофия Дюшенна: рандомизированное контролируемое исследование преднизона (18 месяцев) и азатиоприн (12 месяцев) Неврология. 1993; 43: 520–527. [PubMed] [Google Scholar] 17. Киршнер Дж., Шессл Дж., Шара Ю. и др. Лечение мышечной ткани Дюшенна дистрофия с циклоспорином А: рандомизированный, двойной слепой, плацебо- контролируемое многоцентровое исследование. Lancet Neurol. 2010; 9: 1053–1059. [PubMed] [Google Scholar] 18. Mendell JR, Rodino-Klapac LR, Malik V. Молекулярная терапия стратегии, направленные на мышечную дистрофию Дюшенна.J Child Neurol. 2010; 25: 1145–1148. [Бесплатная статья PMC] [PubMed] [Google Scholar] 19. Arechavala-Gomeza V, Kinali M, Feng L, et al. Ревертантные волокна и следы дистрофина при мышечной дистрофии Дюшенна: значение для клинические испытания. Нервно-мышечное расстройство. 2010; 20: 295–301. [PubMed] [Google Scholar] 20. Лу QL, Rabinowitz A, Chen YC, et al. Системная доставка антисмысловой олигорибонуклеотид восстанавливает экспрессию дистрофина во всем теле скелетные мышцы. Proc Natl Acad Sci USA. 2005. 102: 198–203. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21.Deutekom JC, Janson AA, Ginjaar IB, et al. Местный дистрофин восстановление антисмысловым олигонуклеотидом PRO051. N Engl J Med. 2007; 357: 2677–2686. [PubMed] [Google Scholar] 22. Гоэманс Н.М., Тулиниус М., Аккер Дж. Т. и др. Системное введение PRO051 при мышечной дистрофии Дюшенна. N Engl J Med. 2011; 364: 1513–1522. [PubMed] [Google Scholar] 24. Moulton HM, Moulton JD. Морфолино и их пептидные конъюгаты: терапевтические перспективы и вызов для мышечной дистрофия. Biochim Biophys Acta.2010; 1798: 2296–2303. [PubMed] [Google Scholar] 25. Malerba A, Sharp PS, Graham IR, et al. Хроническая системная терапия с низкими дозами морфолиноолигомеров улучшает патологию и нормализует двигательное поведение у мышей MDX. Mol Ther. 2011; 19: 345–354. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Кинали М., Аречавала-Гомеза В., Фенг Л. и др. Местная реставрация экспрессии дистрофина с морфолиноолигомером AVI-4658 при мышечной дистрофии Дюшенна: простой слепой, плацебо-контролируемый, эскалация дозы, исследование, подтверждающее правильность концепции.Lancet Neurol. 2009; 8: 918–928. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Нери М., Торелли С., Браун С. и др. Уровень дистрофина всего 30% достаточны, чтобы избежать мышечной дистрофии у человека. Нейромускул Disord. 2007; 17: 913–918. [PubMed] [Google Scholar] 28. Политано Л., Нигро Г., Нигро В. и др. Введение гентамицина в Пациенты Дюшена с преждевременным стоп-кодоном. Предварительные результаты. Acta Myol. 2003; 22: 15–21. [PubMed] [Google Scholar] 29. Вагнер К.Р., Хамед С., Хэдли Д.В. и др. Лечение гентамицином Мышечная дистрофия Дюшенна и Беккера из-за бессмысленных мутаций.Энн Нейрол. 2001; 49: 706–711. [PubMed] [Google Scholar] 30. Малик В., Родино-Клапац Л. Р., Виоллет Л. и др. Гентамицин-индуцированный определение стоп-кодонов при мышечной дистрофии Дюшенна. Аня Neurol. 2010. 67: 771–780. [PubMed] [Google Scholar] 31. Финкель Р., Вонг Б., Бушби К. и др. Результаты Фазы 2b, исследования дозирования аталурена (PTC124®) при нейромышечном расстройстве с нонсенс-мутацией Дюшенна / Беккера (nmDBMD). 2010. 20: 656–656. [Google Scholar] 32. Вагнер К. Р., Флекенштейн Дж. Л., Амато А. А. и др.Испытание фазы I / II MYO-029 у взрослых с мышечной дистрофией. Энн Нейрол. 2008; 63: 561–571. [PubMed] [Google Scholar] 33. Chaubourt E, Fossier P, Baux G, et al. Херегулин улучшает состояние дистрофический фенотип у мышей MDX. Acad Sci USA. 2004. 101: 13856–13860. [Бесплатная статья PMC] [PubMed] [Google Scholar] 34. Krag TO, Bogdanovich S, Jensen CJ, et al. Херегулин улучшает состояние дистрофический фенотип у мышей MDX. Proc Natl Acad Sci USA. 2004. 101: 13856–13860. [Бесплатная статья PMC] [PubMed] [Google Scholar] 35.Куинливан Р., Ропер Х., Дэви М. и др. Отчет о мышечной дистрофии Семинар, финансируемый кампанией, Бирмингем, Великобритания, январь 16 2004 г. Остеопороз при мышечной дистрофии Дюшенна; это распространенность, лечение и профилактика. Нервно-мышечное расстройство. 2005; 15: 72–79. [PubMed] [Google Scholar] 36. Бахрах Л.К. Меры по уменьшению остеопороза у Дюшенна мышечная дистрофия. Нервно-мышечное расстройство. 2005. 15: 86–87. [PubMed] [Google Scholar]Медикаментозное лечение мышечной ткани Дюшенна имеющиеся данные и перспективы
Acta Myol.2012 May; 31 (1): 4–8.
Отделение нейропедиатрии и мышечных заболеваний, Университетский медицинский центр Фрайбурга, Германия
Адрес для корреспонденции: Янбернд Киршнер, Отделение нейропедиатрии и мышечных заболеваний, Университетский медицинский центр Фрайбурга, Mathildenstrasse 1, 79106 Freiburg, Германия. Электронное письмо: [email protected] Авторское право Журнал и отдельные статьи, содержащиеся в нем, защищены авторским правом Академии Гаэтано Конте, Неаполь, Италия Это статья в открытом доступе, распространяемая в соответствии с условиями некоммерческой лицензии Creative Commons Attribution Non-Derivatives License, которая разрешает некоммерческое использование, распространение и воспроизведение на любом цифровом носителе при условии, что оригинальная работа должным образом процитирована и не будет изменена в каких-либо способ.Для получения подробной информации, пожалуйста, обратитесь к http://creativecommons.org/licenses/by-nc-nd/3.0/Эта статья цитируется другими статьями в PMC.Abstract
Мышечная дистрофия Дюшенна (МДД) — это заболевание, связанное с Х-хромосомой, которое поражает 1 из 3 600-6 000 новорожденных мальчиков. Это проявляется в отсутствии белка дистрофина в мышечных волокнах, который вызывает прогрессирующее повреждение, ведущее к смерти на третьем десятилетии жизни. Единственными лекарствами, которые до сих пор доказали свою эффективность в замедлении прогрессирования этого заболевания, являются кортикостероиды, которые, как было показано в рандомизированных контролируемых исследованиях, увеличивают мышечную силу; Долгосрочные исследования показали, что они увеличивают время ходьбы и замедляют прогрессирование респираторной дисфункции, дилатационной кардиомиопатии и сколиоза.Несколько потенциальных лекарств сейчас исследуются. Генетическая терапия, включающая введение гена дистрофина через вектор, доказала свою эффективность на животных, но не на людях. В настоящее время проходит клиническое исследование Аталурена, молекулы, которая связывается с рибосомами и может допускать вставку аминокислоты в кодон преждевременной терминации и пропуск экзона, который связывается с РНК и исключает определенные сайты сплайсинга РНК, производя дистрофин меньшего размера. но функциональный. Есть также исследования, пытающиеся модулировать другие мышечные белки, такие как миостатин и атрофин, для уменьшения симптомов.В этой статье не рассматривается лечение кардиомиопатии у пациентов с МДД.
Ключевые слова: мышечная дистрофия Дюшенна, медикаментозное лечение, клиника. испытания
Введение
Мышечная дистрофия Дюшенна (МДД) — это заболевание, связанное с Х-хромосомой, поражающее 1 из 3 600-6 000 новорожденных мальчиков. Он характеризуется слабостью проксимальных мышц, выражающейся в положительном знаке Гауэрса при вставании, аномальной походкой, гипертрофией икроножных мышц и повышенным уровнем креатинкиназы.Большинству пациентов диагноз ставится в возрасте 5 лет, когда симптомы становятся более очевидными. Заболевание имеет прогрессирующее течение мышечной слабости, поражающей также сердечные мышцы и дыхательную систему. Больные мальчики обычно прекращают ходить в возрасте 13 лет и, если их не лечить, умирают до 20 лет от сердечных заболеваний или респираторных инфекций (1, 2).
DMD вызывается мутациями в гене дистрофина, что приводит к серьезному снижению или полному отсутствию белка дистрофина, который обнаруживается в сарколемме мышечных волокон и состоит из четырех частей: C-концевого домена, который присоединяется к другим белкам в мышечных волокнах. мембрана, называемая комплексом дистрогликана, стержневым доменом, богатым цистеином доменом и N-концевым доменом, который связывается с актином (3).Таким образом, С-концевой домен взаимодействует как мост между сарколеммой и внеклеточным матриксом и связывается с другими мембранными белками через дистрофин-гликопротеиновый комплекс. Предполагается, что дистрофин необходим для преобразования силы сократительного аппарата во внеклеточный матрикс и защищает мышечные волокна от любого повреждения, вызванного сокращением мышц, которое может привести к некрозу (3, 4). Отсутствие дистрофина приводит к механическому повреждению сарколеммы, потере гомеостаза кальция и прогрессирующей дегенерации мышечных волокон.
Ген дистрофина — самый крупный ген, обнаруженный у человека, и на его долю приходится примерно 0,1% всего генома человека. Он расположен на коротком плече Х-хромосомы и состоит из 79 экзонов и 7 промоторных областей (4). Сообщаемая частота различных мутаций, приводящих к МДД, широко варьируется. По данным базы данных Лейдена (www.dmd.nl), дупликации одного или нескольких экзонов соответствуют 7% мутаций, точечные мутации составляют 20%, а делеции наблюдаются у 72% пациентов (4, 5).Большинство делеций происходит между экзонами 44 и 55, соответствующими стержневому домену дистрофина (6). Если эти мутации изменяют рамку считывания дистрофина (вне рамки мутации), образование белка прекращается, дистрофин не вырабатывается и у пациента развивается МДД. Если мутация находится «в рамке», дистрофин меньше по размеру, но все еще функционирует, и в этом случае пациенту ставится диагноз мышечной дистрофии Беккера (7).
Хотя молекулярное происхождение МДД известно уже несколько лет, до сих пор не существует лечебного лечения этого заболевания.На сегодняшний день единственным эффективным средством замедления прогрессирования заболевания являются кортикостероиды. Они изменили естественное течение МДД. Однако их точный механизм действия полностью не изучен (2).
Лекарственные препараты
Кортикостероиды: преднизон и дефлазакорт
Глюкокортикоиды, точнее преднизон и дефлазакорт, являются основными лекарственными средствами для лечения МДД. Они используются уже более двух десятилетий, и теперь их преимущества хорошо известны.Это единственное лекарство, которое увеличивает мышечную силу. Ранние исследования доказали, что их использование продлевает ходьбу и улучшает их функциональность в повседневной деятельности. Долгосрочные исследования показали, что они также уменьшают потребность в хирургии сколиоза, улучшают функцию легких и помогают поддерживать сердечную функцию (8, 9).
В 2005 г. Американская академия неврологии, а в 2008 г. — Кокрановский обзор, оценили все рандомизированные контролируемые испытания по применению кортикостероидов и пришли к выводу, что преднизон вводят в дозах 0.75 мг / кг / день показали увеличение мышечной силы и улучшение результатов в стандартизированных функциональных тестах в краткосрочной перспективе (10, 11). Увеличение мышечной силы происходит в течение первых шести месяцев лечения, после чего следует период стабилизации в течение двух лет с последующим снижением, которое происходит медленнее, чем у нелеченных пациентов (12).
В пяти недавно опубликованных долгосрочных контролируемых нерандомизированных исследованиях (продолжительностью более 3 лет) с преднизоном или дефлазакортом сообщалось, что после приема одного из этих препаратов пациенты могут ходить на 2-5 лет дольше, чем те, кто не получает кортикостероиды, необходимость стабилизации позвоночника. количество операций сократилось, а потребность в неинвазивной вентиляции легких отпала (8).Два из этих исследований оценивали сердечную функцию и продемонстрировали, что фракция выброса левого желудочка у пролеченных пациентов была значительно лучше сохранена по сравнению с нелеченными пациентами (8). Другое исследование показало, что 93% пациентов, получавших преднизолон, не имели желудочковой дисфункции в возрасте 12 лет по сравнению с 53% пациентов, не получавших лечения (13). Исследования также показывают увеличение продолжительности жизни (14). Игл упоминает, что в 1960 году средняя продолжительность жизни пациентов с МДД составляла 14 лет.4 года, которые к 1990 году выросли до 19,3 года с использованием кортикостероидов, антибиотикотерапии и интенсивной терапии. В настоящее время он увеличился до 24,5 лет, вероятно, из-за использования неинвазивной вентиляции (8, 14).
Таким образом, есть существенные доказательства, позволяющие рекомендовать использование кортикостероидов всем пациентам с МДД с целью сохранения времени ходьбы как можно дольше и уменьшения легочных, сердечных и ортопедических осложнений (2). Естественно, следует учитывать и побочные эффекты лечения кортикостероидами.Наиболее частым побочным эффектом при длительном лечении является уменьшение роста пациента. Увеличение веса является второй по частоте, но основной причиной прекращения лечения (8). Увеличение веса у пациентов с МДД, получающих стероидные препараты, является многофакторной проблемой. Это не только побочный эффект кортикостероидов — это также результат их ограниченной подвижности, поскольку увеличение веса обычно более выражено у неамбулаторных пациентов. Дефлазакорт вызывал меньшую прибавку в весе, но больше катаракт, чем преднизон.Частота переломов позвонков у пролеченных пациентов варьировала на 5-32%. Около 80% этих переломов были обнаружены при рутинных рентгенологических исследованиях сколиоза и не сопровождались клиническими симптомами (8, 15). К другим побочным эффектам относятся кушингоидная фация, угри, гирсутизм, артериальная гипертензия, расстройство поведения, задержка полового созревания, переломы позвонков, иммуносупрессия и желудочно-кишечные проблемы. Хотя эти побочные эффекты менее часты при использовании в указанных дозах, тем не менее, они являются условиями, которые необходимо контролировать.
Указанная доза преднизона составляет 0,75 мг / кг в сутки. Дозы менее 0,3 мг / кг менее эффективны, а ежедневное введение кажется более эффективным, чем через день (10).
Прерывистые режимы постулируются как имеющие лучший профиль безопасности с точки зрения побочных эффектов, но данные единственного рандомизированного контролируемого исследования прерывистого преднизона были недоступны (что позволило бы провести статистический анализ и сравнение с другими режимами). В одном открытом когортном исследовании с периодическим применением дефлазакорта сообщалось о продлении передвижения (12).Следовательно, необходимы рандомизированные контролируемые исследования для определения оптимального графика лечения в долгосрочной перспективе.
В настоящее время рекомендуется начинать лечение, когда пациент находится в «фазе плато». Обычно это происходит в возрасте от 4 до 6 лет, когда мальчик перестает двигать вперед. В настоящее время многие врачи продолжают лечение даже после того, как пациент с МДД потерял способность ходить, с целью сохранения функции верхних конечностей, снижения скорости прогрессирования сколиоза и замедления нарушения дыхательной и сердечной функции ( 2).По-прежнему необходимы дальнейшие исследования, чтобы определить, продолжают ли неамбулаторные пациенты получать пользу от этого лечения.
Дефлазакорт представляет собой оксазолиновое производное преднизона и в дозе 0,9 мг / кг / день так же эффективен, как и преднизон, при лечении МДД. Выбор зависит от местной доступности дефлазакорта и преднизона, его стоимости, состава и предпочтений пациента. Одно небольшое рандомизированное исследование предполагает более высокую частоту и выраженность увеличения веса при приеме преднизона, чем при применении дефлазакорта (13).Таким образом, некоторые пациенты предпочитают дефлазакорт. Однако риск развития катаракты повышен по сравнению с преднизоном: в нерандомизированных исследованиях 10-30% катаракт наблюдались в среднем через 3,2 года лечения дефлазакортом. Таким образом, эти пациенты должны ежегодно наблюдаться офтальмологом (2, 12).
Другие иммунодепрессанты
Было высказано предположение, что польза от лечения кортикостероидами может быть связана с иммунодепрессивным эффектом.Таким образом, были проведены дальнейшие исследования с другими иммунодепрессантами. Однако рандомизированное контролируемое исследование 99 мальчиков с МДД, получавших только азатиприн или в комбинации с преднизоном, показало, что азатиоприн не имеет положительного эффекта (16). Плацебоконтролируемое двойное слепое исследование 146 амбулаторных пациентов с МДД, получавших только циклоспорин-А или плацебо в сочетании с преднизоном, не выявило разницы в силе мышц и функциональных возможностях между группами лечения (17).
Генетическая терапия
Недавно были проведены исследования возможностей генетической терапии, посредством которой встраивается ген дистрофина. Однако на этом пути встретилось несколько препятствий. Размер гена дистрофина затрудняет работу с ним в генной терапии. Таким образом, были разработаны гены меньшего размера, микро- или мини-дистрофин, которые можно вставить в вектор. Наиболее подходящим вектором, обнаруженным на данный момент, является вирус, связанный с аденовирусом, непатогенным парвовирусом, но было показано, что он вызывает иммунологический ответ.Чтобы оценить ответ, были созданы mdx мышей dys- / dys-, и есть свидетельства того, что при инъекции гена дистрофин частично экспрессируется, а мышечная сила повышается. Однако в предварительных исследованиях на людях через 90 дней после начала лечения экспрессия этого гена не наблюдалась. Результаты показывают, что клеточный иммунитет препятствует успеху этой терапии (18, 19).
Пропуск экзона
При мышечной дистрофии Беккера белок дистрофина меньше по размеру и частично функционирует, что приводит к менее тяжелому заболеванию.У пациентов с МДД, как упоминалось ранее, мутировавший ген проявляет делеции, дупликации и точечные мутации, которые нарушают рамку считывания генетической информации. В настоящее время исследователи стремятся ввести молекулу, способную вмешиваться в сигналы сплайсинга РНК, чтобы пропустить дополнительный соседний экзон, тем самым восстанавливая рамку считывания и обеспечивая экспрессию белка меньшего размера, но частично функционального, как у пациентов, у которых есть Мышечная дистрофия Беккера. Эта синтетическая модифицированная молекула РНК называется антисмысловым олигонуклеотидом (АО) и способна связываться со специфическими участками пре-РНК, маскируя и исключая этот экзон из сплайсинга (5).Эта терапия будет индивидуальной для каждой мутации. Текущие исследования пропуска экзона направлены на пропуск экзона 51, «пропуск» которого применим к самой большой группе пациентов, включающей 13% всех пациентов с МДД, за которыми следуют мутации в экзоне 45. В настоящее время изучаются два АО:
2′-O-метилфосфориоаты (2OMP): исследования первоначально проводились на мышах mdx с 2OMP на экзоне 23. Они показали присутствие дистрофина во многих волокнах скелетных мышц, но не в сердце (20). .Затем было проведено клиническое исследование четырех пациентов с МДД с PRO051 / GSK2402964 (2OMP, нацеленным на экзон 51), и исследователи обнаружили, что 64-97% мышечных волокон экспрессируют белок дистрофин в количестве 17-35%. Побочных эффектов не наблюдалось (21). В настоящее время проводится многоцентровое исследование (22, 23).
Морфолиноолигомер фофородиамидата (PMO): исследования на мышах MDX показали присутствие дистрофина не только в мышечных волокнах, но и в сердце после введения высоких доз препарата.Длительное повторяющееся лечение даже продемонстрировало улучшение двигательной функции (24). На людях исследование экзона 51 у семи пациентов с МДД (AVI-4658) показало, что те, кто получал высокие дозы (0,9 мг), продуцировали дистрофин на 22-32% нормальных уровнях в 44-79% мышечных волокон. В этом исследовании не наблюдалось никаких признаков токсичности (25), в то время как предыдущее, проведенное на нечеловеческих приматах, показало дегенерацию канальцев в почках (26). Недостатком этого препарата является то, что эффект носит временный характер и ограничен временем, в течение которого АО остается в ткани.Кроме того, он производит уровни дистрофина ниже 30-60% от нормы, что, как предполагается, достаточно для компенсации мышечной дисфункции (27).
Преждевременное считывание стоп-кодона: аминогликозиды и аталурен
Эта форма лечения применяется только к мальчикам с мутациями, приводящими к преждевременным кодонам терминации, которые, по оценкам, встречаются примерно у 13-15% популяции МДД. Было показано, что считывание стоп-кодона способно подавлять кодоны терминации за счет неправильного считывания РНК, тем самым позволяя вставку альтернативных аминокислот в сайт мутировавшего кодона преждевременной терминации.В результате образуется полноразмерный белок-дистрофин с заменой только одной аминокислоты.
Аминогликозиды
В культурах клеток гентамицин взаимодействует с 40-й рибосомной субъединицей при транскрипции РНК, подавляя терминальные кодоны и вставляя на их место другую заменяющую ее аминокислоту. В исследованиях на мышах mdx и на людях гентамицин был способен вызывать экспрессию дистрофина в мышечных волокнах на 20% от нормального уровня (5). Однако исследования пациентов с МДД остаются противоречивыми.Фактически одно из них показало положительный эффект мышечной силы и повторную экспрессию дистрофина в мышцах (28), в то время как в другом исследовании с участием 12 пациентов с МДД, проведенном в течение шести месяцев, экспрессия дистрофина была обнаружена только в 6 из них. у 12 пациентов, и никаких клинических преимуществ не наблюдалось (29-30).
Аталурен (PTC124)
Аталурен — это пероральный препарат, который теоретически имеет тот же эффект, что и гентамицин, но который связан с 60-й рибосомной субъединицей.Его эффективность у мышей mdx аналогична гентамицину, вызывая экспрессию дистрофина в 20-25% мышечных волокон (5).
С этими результатами было проведено двойное слепое рандомизированное многоцентровое исследование с участием 174 пациентов. После 48 недель приема низких доз аталурена пациенты показали некоторое улучшение в тесте с 6-минутной ходьбой, но окончательные результаты не были опубликованы.
Миостатин
Миостатин — это белок, входящий в семейство трансформирующих факторов роста B, регулирующих размер мышц.Поскольку мышечная гипертрофия наблюдалась у мышей с нокаутом гена миостатина ( mys- / mys- ), блокировка миостатина может служить потенциальным средством лечения МДД. Было предложено несколько способов блокирования миостатина: фолластатин, блокатор рецепторов миостатина и деструктивный пропуск экзона для гена миостатина (5).
Антитело миостатина вводили мышам MDX, вызывая мышечную гипертрофию и большую силу. Только одно клиническое исследование было проведено на людях с антителом против миостатина (MYO-029) у взрослых пациентов с мышечной дистрофией, которое хорошо переносилось, но не увеличивало мышечную силу (32).
Утрофин
Утрофин разделяет 80% последовательности дистрофина и сверхэкспрессируется в мышцах пациентов с МДД. Было высказано предположение, что его повышенная регуляция может замедлить прогрессирование болезни. Инъекция херегулина, ларгинина или оксида азота мышам MDX увеличивает экспрессию утрофина в гистологических препаратах (33), а внутрибрюшинное введение белка TAT-утрофина увеличивает мышечную силу у крыс MDX. Было проведено исследование на здоровых людях с использованием препарата, разработанного Summit PLC (C110 / BM195), который оказался хорошо переносимым, хотя фармакокинетика препарата не позволяла продолжить лечение (5).
Добавка витамина D
Пациенты с МДД имеют повышенный риск развития патологических переломов длинных костей и позвоночника. Во-первых, минеральная плотность костной ткани снижается как следствие ограниченной подвижности. Во-вторых, риск остеопороза увеличивается из-за хорошо известного побочного эффекта длительного лечения кортикостероидами. В-третьих, они, как правило, имеют низкий уровень витамина D, вероятно, из-за меньшего воздействия солнечного света. Поэтому рекомендуются физические упражнения и достаточное количество кальция и витамина D в рационе.Добавки витамина D рекомендуется пациентам с доказанным дефицитом витамина D (сывороточный 25-гидроксивитамин D <50 мг / мл) (2, 35).
Нет данных о том, что бисфосфаты следует использовать с профилактической целью у детей, получающих стероидную терапию, однако введение этого препарата рекомендуется при возникновении патологических переломов (7, 35, 36).
Заключение
Хотя молекулярная основа мышечной дистрофии Дюшенна известна уже несколько десятилетий, до сих пор не существует лечения, которое излечивает эту болезнь.В настоящее время ведутся активные исследования по этой теме. До сих пор единственным лекарством, способным замедлить прогрессирование этого заболевания, были кортикостероиды.
Список литературы
1. Пойски Дж. Паттерны поведения при мышечной дистрофии Дюшенна: отчет на семинаре по поведению «Мышечная дистрофия» родительского проекта 8-9 декабря 2006 г., Филадельфия, США. Нервно-мышечное расстройство. 2007; 17: 986–994. [PubMed] [Google Scholar] 2. Бушби К., Финкель Р., Бирнкрант Д. Д. и др. Диагностика и лечение мышечной дистрофии Дюшенна, часть 1: диагностика и фармакологическое и психосоциальное управление.Lancet Neurol. 2010; 9: 77–93. [PubMed] [Google Scholar] 3. О’Брайен К.Ф., Кункель Л.М. Дистрофин и мышечная дистрофия: в прошлом, настоящее и будущее. Mol Genet Metab. 2001. 74: 75–88. [PubMed] [Google Scholar] 4. Мунтони Ф., Торелли С., Ферлини А. и др. Дистрофин и мутации: один ген, несколько белков, несколько фенотипов. Lancet Neurol. 2003; 2: 731–740. [PubMed] [Google Scholar] 5. Пичавант С., Аартсма-Рус А., Клеменс П.Р. и др. Текущее состояние фармацевтические и генетические терапевтические подходы к лечению МДД.2011. Mol Ther. 2011; 19: 830–840. [Бесплатная статья PMC] [PubMed] [Google Scholar] 6. Нельсон С.Ф., Кросби Р., Мичели М.К. и др. Новые генетические методы лечения для лечения мышечной дистрофии Дюшенна. Curr Opin Neurol. 2009; 22: 532–538. [Бесплатная статья PMC] [PubMed] [Google Scholar] 7. Манзур А.Ю., Кинали М., Мунтони Ф. Последние сведения об управлении Мышечная дистрофия Дюшенна. Arch Dis Child. 2008; 93: 986–990. [PubMed] [Google Scholar] 8. Moxley RT, Pandya S, Ciafoli E, et al. Изменения в естественной истории мышечной дистрофии Дюшенна при длительном приеме кортикостероидов лечение: значение для управления.J Child Neurol. 2010; 25: 1116–1129. [PubMed] [Google Scholar] 9. Houde S, Filiatrault M, Fournier A и др. Использование дефлазакорта при Дюшенне мышечная дистрофия: наблюдение через 8 лет. Pediatr Neurol. 2008. 38: 200–206. [PubMed] [Google Scholar] 10. Moxley RT, 3rd, Ashwal S, Pandya S, et al. Параметр практики: кортикостероид лечение дистрофии Дюшенна: отчет о качестве Подкомитет по стандартам Американской академии неврологии и Практический комитет Общества детской неврологии. Неврология.2005; 64: 13–20. [PubMed] [Google Scholar] 11. Анджелини Дж. Роль кортикостероидов при мышечной дистрофии: а Критическая оценка. Мышечный нерв. 2007. 36: 424–435. [PubMed] [Google Scholar] 12. Манзур А.Ю., Кунцер Т., Пайк М. и др. Глюкокортикоидные кортикостероиды при мышечной дистрофии Дюшенна. Кокрановская база данных Syst Ред. 2008 г .; 1: CD003725 – CD003725. [PubMed] [Google Scholar] 13. Маркхэм Л.В., Киннетт К., Вонг Б.Л. и др. Кортикостероидное лечение замедляет развитие желудочковой дисфункции в мышечной ткани Дюшенна. дистрофия.Нервно-мышечное расстройство. 2008. 18: 365–370. [PubMed] [Google Scholar] 14. Игл М., Бурк Дж., Баллок Р. и др. Управление мышечной массой Дюшенна дистрофия — аддитивный эффект хирургии позвоночника и домашнего ночная вентиляция для повышения выживаемости. Нервно-мышечное расстройство. 2007. 17: 470–475. [PubMed] [Google Scholar] 15. King WM, Ruttencutter R, Nagaraja HN и др. Ортопедические исходы длительного ежедневного лечения кортикостероидами при Дюшенне мышечная дистрофия. Неврология. 2007. 68: 1607–1613. [PubMed] [Google Scholar] 16.Griggs RC, Moxley RT, 3rd, Mendell JR, et al. Дистрофия Дюшенна: рандомизированное контролируемое исследование преднизона (18 месяцев) и азатиоприн (12 месяцев) Неврология. 1993; 43: 520–527. [PubMed] [Google Scholar] 17. Киршнер Дж., Шессл Дж., Шара Ю. и др. Лечение мышечной ткани Дюшенна дистрофия с циклоспорином А: рандомизированный, двойной слепой, плацебо- контролируемое многоцентровое исследование. Lancet Neurol. 2010; 9: 1053–1059. [PubMed] [Google Scholar] 18. Mendell JR, Rodino-Klapac LR, Malik V. Молекулярная терапия стратегии, направленные на мышечную дистрофию Дюшенна.J Child Neurol. 2010; 25: 1145–1148. [Бесплатная статья PMC] [PubMed] [Google Scholar] 19. Arechavala-Gomeza V, Kinali M, Feng L, et al. Ревертантные волокна и следы дистрофина при мышечной дистрофии Дюшенна: значение для клинические испытания. Нервно-мышечное расстройство. 2010; 20: 295–301. [PubMed] [Google Scholar] 20. Лу QL, Rabinowitz A, Chen YC, et al. Системная доставка антисмысловой олигорибонуклеотид восстанавливает экспрессию дистрофина во всем теле скелетные мышцы. Proc Natl Acad Sci USA. 2005. 102: 198–203. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21.Deutekom JC, Janson AA, Ginjaar IB, et al. Местный дистрофин восстановление антисмысловым олигонуклеотидом PRO051. N Engl J Med. 2007; 357: 2677–2686. [PubMed] [Google Scholar] 22. Гоэманс Н.М., Тулиниус М., Аккер Дж. Т. и др. Системное введение PRO051 при мышечной дистрофии Дюшенна. N Engl J Med. 2011; 364: 1513–1522. [PubMed] [Google Scholar] 24. Moulton HM, Moulton JD. Морфолино и их пептидные конъюгаты: терапевтические перспективы и вызов для мышечной дистрофия. Biochim Biophys Acta.2010; 1798: 2296–2303. [PubMed] [Google Scholar] 25. Malerba A, Sharp PS, Graham IR, et al. Хроническая системная терапия с низкими дозами морфолиноолигомеров улучшает патологию и нормализует двигательное поведение у мышей MDX. Mol Ther. 2011; 19: 345–354. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Кинали М., Аречавала-Гомеза В., Фенг Л. и др. Местная реставрация экспрессии дистрофина с морфолиноолигомером AVI-4658 при мышечной дистрофии Дюшенна: простой слепой, плацебо-контролируемый, эскалация дозы, исследование, подтверждающее правильность концепции.Lancet Neurol. 2009; 8: 918–928. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Нери М., Торелли С., Браун С. и др. Уровень дистрофина всего 30% достаточны, чтобы избежать мышечной дистрофии у человека. Нейромускул Disord. 2007; 17: 913–918. [PubMed] [Google Scholar] 28. Политано Л., Нигро Г., Нигро В. и др. Введение гентамицина в Пациенты Дюшена с преждевременным стоп-кодоном. Предварительные результаты. Acta Myol. 2003; 22: 15–21. [PubMed] [Google Scholar] 29. Вагнер К.Р., Хамед С., Хэдли Д.В. и др. Лечение гентамицином Мышечная дистрофия Дюшенна и Беккера из-за бессмысленных мутаций.Энн Нейрол. 2001; 49: 706–711. [PubMed] [Google Scholar] 30. Малик В., Родино-Клапац Л. Р., Виоллет Л. и др. Гентамицин-индуцированный определение стоп-кодонов при мышечной дистрофии Дюшенна. Аня Neurol. 2010. 67: 771–780. [PubMed] [Google Scholar] 31. Финкель Р., Вонг Б., Бушби К. и др. Результаты Фазы 2b, исследования дозирования аталурена (PTC124®) при нейромышечном расстройстве с нонсенс-мутацией Дюшенна / Беккера (nmDBMD). 2010. 20: 656–656. [Google Scholar] 32. Вагнер К. Р., Флекенштейн Дж. Л., Амато А. А. и др.Испытание фазы I / II MYO-029 у взрослых с мышечной дистрофией. Энн Нейрол. 2008; 63: 561–571. [PubMed] [Google Scholar] 33. Chaubourt E, Fossier P, Baux G, et al. Херегулин улучшает состояние дистрофический фенотип у мышей MDX. Acad Sci USA. 2004. 101: 13856–13860. [Бесплатная статья PMC] [PubMed] [Google Scholar] 34. Krag TO, Bogdanovich S, Jensen CJ, et al. Херегулин улучшает состояние дистрофический фенотип у мышей MDX. Proc Natl Acad Sci USA. 2004. 101: 13856–13860. [Бесплатная статья PMC] [PubMed] [Google Scholar] 35.Куинливан Р., Ропер Х., Дэви М. и др. Отчет о мышечной дистрофии Семинар, финансируемый кампанией, Бирмингем, Великобритания, январь 16 2004 г. Остеопороз при мышечной дистрофии Дюшенна; это распространенность, лечение и профилактика. Нервно-мышечное расстройство. 2005; 15: 72–79. [PubMed] [Google Scholar] 36. Бахрах Л.К. Меры по уменьшению остеопороза у Дюшенна мышечная дистрофия. Нервно-мышечное расстройство. 2005. 15: 86–87. [PubMed] [Google Scholar]FDA одобрило целевое лечение редкой мутации мышечной дистрофии Дюшенна
- Для немедленного выпуска:
Сегодня U.Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США предоставило ускоренное разрешение на инъекцию Вилтепсо (вилтоларсена) для лечения мышечной дистрофии Дюшенна (МДД) у пациентов с подтвержденной мутацией гена МДД, поддающейся пропуску экзона 53. Это второе целевое лечение, одобренное FDA для пациентов с этим типом мутации. Примерно 8% пациентов с МДД имеют мутацию, которая поддается пропуску экзона 53.
«FDA стремится способствовать разработке лекарств от серьезных неврологических расстройств, таких как мышечная дистрофия Дюшенна», — сказал Билли Данн, M.D., директор отдела неврологии Центра оценки и исследований лекарственных средств FDA. «Сегодняшнее одобрение Viltepso обеспечивает важный вариант лечения пациентов с мышечной дистрофией Дюшенна с этой подтвержденной мутацией».
МДД — редкое генетическое заболевание, характеризующееся прогрессирующим разрушением мышц и слабостью. Это наиболее распространенный тип мышечной дистрофии. МДД вызывается мутациями в гене МДД, что приводит к отсутствию дистрофина, белка, который помогает сохранить целостность мышечных клеток.Первые симптомы обычно проявляются в возрасте от трех до пяти лет и со временем ухудшаются. МДД встречается примерно у одного из 3600 младенцев мужского пола во всем мире; в редких случаях может поражать самок.
Viltepso оценивался в двух клинических исследованиях с участием 32 пациентов, все из которых были мужчинами с генетически подтвержденным МДД. Увеличение выработки дистрофина было установлено в одном из этих двух исследований, в котором участвовали 16 пациентов с МДД, из которых 8 пациентов получали Вилтепсо в рекомендованной дозе.В исследовании уровень дистрофина увеличился в среднем с 0,6% от нормы на исходном уровне до 5,9% от нормы на 25 неделе.
FDA пришло к выводу, что данные заявителя продемонстрировали увеличение выработки дистрофина, что с достаточной вероятностью предсказывает клиническую пользу у пациентов с МДД, у которых есть подтвержденная мутация гена дистрофина, поддающаяся пропуску экзона 53. Клиническая польза препарата не установлена. Принимая это решение, FDA учло потенциальные риски, связанные с препаратом, опасный для жизни и изнурительный характер заболевания, а также отсутствие доступных методов лечения.
В рамках ускоренного процесса утверждения FDA требует, чтобы компания провела клинические испытания для подтверждения клинической пользы препарата. Текущее исследование предназначено для оценки того, улучшает ли Viltepso время простоя у пациентов с МДД с этой подтвержденной мутацией. Если в ходе исследования не удается подтвердить клиническую пользу, FDA может инициировать процедуру отмены одобрения препарата.
Наиболее частыми побочными эффектами, наблюдаемыми у пациентов с МДД (объединенные из двух исследований), получавших 80 мг / кг один раз в неделю, были: инфекция верхних дыхательных путей, реакция в месте инъекции, кашель и лихорадка.
Хотя почечная токсичность не наблюдалась в клинических исследованиях Вилтепсо, клинический опыт применения Вилтепсо ограничен, и после введения некоторых антисмысловых олигонуклеотидов наблюдалась почечная токсичность, включая потенциально смертельный гломерулонефрит. У пациентов, принимающих Вилтепсо, следует контролировать функцию почек.
Viltepso был одобрен в рамках ускоренной процедуры одобрения FDA, которая предусматривает одобрение лекарств, которые лечат серьезные или опасные для жизни заболевания и в целом предлагают значительное преимущество перед существующими методами лечения.Одобрение в рамках этого пути может быть основано на адекватных и хорошо контролируемых исследованиях, показывающих, что лекарство влияет на суррогатную конечную точку, которая с достаточной вероятностью предсказывает клиническую пользу для пациентов (то есть, как пациенты чувствуют себя или функционируют или выживают). Этот путь обеспечивает более ранний доступ пациентов к перспективным новым лекарствам, в то время как компания проводит клинические испытания, чтобы проверить прогнозируемую клиническую пользу.
FDA предоставило этой заявке статус приоритетного рассмотрения.
FDA выдает одобрение NS Pharma, Inc.
FDA, агентство в составе Министерства здравоохранения и социальных служб США, защищает здоровье населения, обеспечивая безопасность, эффективность и безопасность лекарственных и ветеринарных препаратов, вакцин и других биологических продуктов для использования человеком, а также медицинских устройств. Агентство также отвечает за безопасность и сохранность продуктов питания, косметики, пищевых добавок, продуктов, излучающих электронное излучение, а также за регулирование табачных изделий.
Связанная информация
###
Симптомы, лечение, типы и причины
Мышечная дистрофия относится к группе заболеваний, которые включают прогрессирующую потерю мышечной массы и, как следствие, потерю силы.
Основные формы мышечной дистрофии могут поражать до 1 из 5000 мужчин.
Самая распространенная форма — мышечная дистрофия Дюшенна. Обычно это поражает маленьких мальчиков, но во взрослом возрасте могут проявиться и другие варианты.
Мышечная дистрофия вызывается генетическими мутациями, которые мешают выработке мышечных белков, необходимых для создания и поддержания здоровых мышц.
Причины генетические. Семейный анамнез мышечной дистрофии увеличивает вероятность того, что она затронет человека.
В настоящее время лекарства нет, но определенные физические и медицинские процедуры могут улучшить симптомы и замедлить прогрессирование.
Краткие сведения о мышечной дистрофии
Вот некоторые ключевые моменты о мышечной дистрофии. Более подробная и вспомогательная информация находится в основной статье.
- Мышечная дистрофия — это совокупность состояний, вызывающих истощение мышц
- Мышечная дистрофия Дюшенна — наиболее распространенный тип
- Недостаток белка, называемого дистрофином, является основной причиной мышечной дистрофии
- В настоящее время проводятся испытания генной терапии для борьбы с ней. болезнь
- В настоящее время нет лекарства от мышечной дистрофии
Мышечная дистрофия — это группа из более чем 30 состояний, которые приводят к мышечной слабости и дегенерации.По мере прогрессирования состояния становится все труднее двигаться. В некоторых случаях это может повлиять на дыхание и работу сердца, что приведет к опасным для жизни осложнениям.
В зависимости от типа и степени тяжести последствия могут быть легкими, медленно прогрессирующими в течение нормальной продолжительности жизни, могут иметь место умеренная инвалидность или привести к летальному исходу.
В настоящее время нет способа предотвратить или обратить вспять мышечную дистрофию, но различные виды терапии и медикаментозного лечения могут улучшить качество жизни человека и замедлить прогрессирование симптомов.
Ниже приведены симптомы мышечной дистрофии Дюшенна, наиболее распространенной формы заболевания.
Симптомы мышечной дистрофии Беккера схожи, но, как правило, начинаются в середине двадцатых годов или позже, менее выражены и прогрессируют медленнее.
Ранние симптомы включают:
- ковыляющую походку
- боль и скованность в мышцах
- трудности с бегом и прыжками
- ходьбу на пальцах ног
- трудности с сидением или стоянием
- нарушения обучаемости, например, развитие речи позже обычное
- частое падение
Со временем становится более вероятным следующее:
- неспособность ходить
- укорочение мышц и сухожилий, дальнейшее ограничение движения
- проблемы с дыханием могут стать настолько серьезными, что потребуется вспомогательное дыхание
- искривление позвоночника может быть вызвано, если мышцы недостаточно сильны, чтобы поддерживать его структуру.
- мышцы сердца могут быть ослаблены, что приводит к сердечным проблемам.
- затрудненное глотание с риском аспирационной пневмонии.Иногда необходима трубка для кормления.
В настоящее время не существует лекарства от мышечной дистрофии. Лекарства и различные методы лечения помогают замедлить прогрессирование заболевания и сохранить подвижность пациента на максимально долгое время.
Лекарства
Два наиболее часто назначаемых лекарства от мышечной дистрофии:
- Кортикостероиды: Этот тип лекарств может помочь увеличить мышечную силу и замедлить прогрессирование, но длительное применение может ослабить кости и увеличить набор веса.
- Сердечные препараты: Если заболевание влияет на сердце, могут помочь бета-блокаторы и ингибиторы ангиотензинпревращающего фермента (АПФ).
Физическая терапия
- Общие упражнения: Диапазон движений и упражнения на растяжку могут помочь бороться с неизбежным движением внутрь конечностей при сокращении мышц и сухожилий. Конечности, как правило, фиксируются в своем положении, и эти виды деятельности могут помочь им дольше оставаться подвижными. Стандартные аэробные упражнения с малой нагрузкой, такие как ходьба и плавание, также могут помочь замедлить прогрессирование болезни.
- Помощь при дыхании: По мере того, как мышцы, используемые для дыхания, становятся слабее, может потребоваться использование устройств для улучшения доставки кислорода в течение ночи. В самых тяжелых случаях пациенту может потребоваться использовать аппарат искусственной вентиляции легких, чтобы дышать от его имени.
- Средства передвижения: Трости, инвалидные коляски и ходунки могут помочь человеку оставаться мобильным.
- Подтяжки: Они удерживают мышцы и сухожилия в напряжении и помогают замедлить их сокращение. Они также оказывают дополнительную поддержку пользователю при перемещении.
Существуют различные типы мышечной дистрофии, в том числе следующие:
- Мышечная дистрофия Дюшенна: Наиболее распространенная форма заболевания. Симптомы обычно появляются до трехлетнего возраста ребенка; они обычно прикованы к инвалидной коляске к 12 годам и умирают от дыхательной недостаточности в возрасте от 20 до 25 лет.
- Мышечная дистрофия Беккера: Симптомы, сходные с симптомами Дюшенна, но с более поздним началом и более медленным прогрессированием; смерть обычно наступает в середине сороковых годов.
- Миотоническая форма (болезнь Штейнерта): Миотоническая форма является наиболее распространенной формой у взрослых. Он характеризуется неспособностью расслабить мышцу после ее сокращения. Часто в первую очередь поражаются мышцы лица и шеи. Симптомы также включают катаракту, сонливость и аритмию.
- Врожденный: Этот тип может быть очевиден с рождения или в возрасте до 2 лет. Поражает девочек и мальчиков. Некоторые формы развиваются медленно, тогда как другие могут двигаться быстро и вызывать значительные нарушения.
- Facioscapulohumeral (FSHD): Начало может быть практически в любом возрасте, но чаще всего встречается в подростковом возрасте. Мышечная слабость часто начинается с лица и плеч. Люди с ЛЛПД могут спать с приоткрытыми глазами и испытывать проблемы с закрытием век. Когда человек с ЛЛПД поднимает руки, его лопатки высовываются, как крылья.
- Конечностный пояс: Этот вариант начинается в детстве или подростковом возрасте и сначала воздействует на мышцы плеча и бедра.Люди с мышечной дистрофией конечностей могут иметь проблемы с поднятием передней части стопы, что делает спотыкание обычной проблемой.
- Глоточная мышечная дистрофия: Начало заболевания в возрасте от 40 до 70 лет. Сначала поражаются веки, горло и лицо, затем плечо и таз.
Мышечная дистрофия вызвана мутациями Х-хромосомы. Каждая версия мышечной дистрофии возникает из-за разного набора мутаций, но все они не позволяют организму вырабатывать дистрофин.Дистрофин — это белок, необходимый для наращивания и восстановления мышц.
Мышечная дистрофия Дюшенна вызывается специфическими мутациями в гене, который кодирует цитоскелетный белок дистрофин. Дистрофин составляет всего 0,002 процента от общего количества белков в поперечно-полосатых мышцах, но это важная молекула для общего функционирования мышц.
Дистрофин является частью невероятно сложной группы белков, которые позволяют мышцам работать правильно. Белок помогает закрепить вместе различные компоненты в мышечных клетках и связывает их все с сарколеммой — внешней мембраной.
Если дистрофин отсутствует или деформирован, этот процесс не работает правильно, и происходят разрывы во внешней мембране. Это ослабляет мышцы, а также может активно повреждать сами мышечные клетки.
При мышечной дистрофии Дюшенна дистрофин почти полностью отсутствует; чем меньше вырабатывается дистрофина, тем хуже симптомы и этиология заболевания. При мышечной дистрофии Беккера наблюдается уменьшение количества или размера белка дистрофина.
Ген, кодирующий дистрофин, является крупнейшим из известных генов человека.Более 1000 мутаций в этом гене были идентифицированы при мышечной дистрофии Дюшенна и Беккера.
Существует множество методов, используемых для окончательной диагностики мышечной дистрофии:
Поделиться на Pinterest Генетические мутации, вызывающие мышечную дистрофию, хорошо известны и могут использоваться для постановки диагноза.- Ферментный анализ: Поврежденные мышцы продуцируют креатинкиназу (СК). Повышенный уровень CK при отсутствии других типов мышечных повреждений может указывать на мышечную дистрофию.
- Генетическое тестирование: Поскольку известно, что при мышечной дистрофии происходят генетические мутации, эти изменения могут быть проверены.
- Мониторинг сердца: Электрокардиография и эхокардиограмма позволяют обнаружить изменения в мускулатуре сердца. Это особенно полезно для диагностики миотонической мышечной дистрофии.
- Мониторинг легких: Проверка функции легких может дать дополнительные доказательства.
- Электромиография: В мышцу вводят иглу для измерения электрической активности.Результаты могут показать признаки мышечного заболевания.
- Биопсия: Удаление части мышцы и ее исследование под микроскопом может выявить явные признаки мышечной дистрофии.
Прогноз будет зависеть от типа мышечной дистрофии и тяжести симптомов.
Мышечная дистрофия Дюшенна может привести к опасным для жизни осложнениям, таким как затрудненное дыхание и проблемы с сердцем.
В прошлом люди с этим заболеванием обычно не доживали до 20 лет, но прогресс улучшает их перспективы.
В настоящее время средняя продолжительность жизни людей с Дюшенном составляет 27 лет, и со временем она может улучшиться по мере продвижения лечения.
Человек с мышечной дистрофией может нуждаться в пожизненной помощи.
Многое известно о механизмах мышечной дистрофии, как мышечной, так и генетической, и хотя полное излечение может быть на некотором расстоянии, есть направления исследований, которые все ближе к нему.
Заместительная генная терапия
Поделиться на PinterestГенная терапия — это лишь одно из направлений исследований в области лечения мышечной дистрофии.Поскольку конкретный ген, участвующий в мышечной дистрофии, был обнаружен, целесообразно рассмотреть замену гена, который мог бы создать недостающий белок дистрофина.
Этот подход сопряжен со сложными проблемами, включая способность иммунной системы отталкивать новый белок и большой размер гена дистрофина, который необходимо заменить. Также существуют трудности с нацеливанием вирусных векторов непосредственно на скелетные мышцы.
Другой подход нацелен на производство атрофина.Утрофин — это белок, похожий на дистрофин, на который не влияет мышечная дистрофия. Если производство атрофина может быть увеличено, болезнь можно остановить или замедлить.
Изменение продукции белка
Если ген дистрофина считывается аппаратом синтеза белка и достигает мутации, он останавливается и не завершает белок. Испытываются лекарства, которые заставляют оборудование для производства белка пропускать мутировавший контент и по-прежнему создавать дистрофин.
Лекарства, замедляющие истощение мышц
Вместо того, чтобы воздействовать на гены, лежащие в основе мышечной дистрофии, некоторые исследователи пытаются замедлить неизбежное истощение мышц.
Мышцы в стандартных условиях могут восстанавливаться сами. Исследования по контролю или увеличению количества таких ремонтов могут показать некоторые преимущества для людей с мышечной дистрофией.
Исследование стволовых клеток
Исследователи изучают возможность введения мышечных стволовых клеток, способных производить недостающий белок дистрофина.
Текущие проекты ищут наиболее полезные для использования типы клеток и способы их доставки в скелетные мышцы.
Трансплантация миобластов
На ранних стадиях мышечной дистрофии миобласты (также называемые сателлитными клетками) восстанавливают и заменяют поврежденные мышечные волокна. По мере истощения миобластов мышцы медленно превращаются в соединительную ткань.
Некоторые исследования пытались внедрить модифицированные клетки миобластов в мышцы, чтобы заменить истощенные естественные миобласты.
Мышечная дистрофия — NHS
Мышечные дистрофии (МД) — это группа наследственных генетических состояний, которые постепенно вызывают ослабление мышц, что приводит к возрастающему уровню инвалидности.
MD — это прогрессирующее состояние, что означает, что со временем оно ухудшается. Он часто начинается с воздействия на определенную группу мышц, а затем затрагивает более широкие мышцы.
Некоторые типы MD в конечном итоге влияют на сердце или мышцы, используемые для дыхания, после чего состояние становится опасным для жизни.
Нет лекарства от MD, но лечение может помочь справиться со многими симптомами.
Что вызывает мышечную дистрофию?
MD вызван изменениями (мутациями) генов, отвечающих за структуру и функционирование мышц человека.
Мутации вызывают изменения в мышечных волокнах, которые мешают мышцам функционировать. Со временем это приводит к увеличению инвалидности.
Мутации часто передаются по наследству от родителей. Если у вас есть семейная история MD, ваш терапевт может направить вас на генетическое тестирование и консультацию, чтобы оценить ваш риск развития этого состояния или рождения ребенка с MD, а также обсудить доступные вам варианты.
Узнайте больше о причинах MD и генетическом тестировании на MD.
Виды мышечной дистрофии
Существует много разных типов MD, каждый из которых имеет несколько разные симптомы. Не все типы вызывают тяжелую инвалидность, и многие не влияют на продолжительность жизни.
Некоторые из наиболее распространенных типов MD включают:
- MD Duchenne MD — одна из самых частых и тяжелых форм, обычно поражает мальчиков в раннем детстве; люди с этим заболеванием обычно доживают до 20-30 лет
- миотоническая дистрофия — разновидность МД, которая может развиться в любом возрасте; На продолжительность жизни не всегда влияет, но люди с тяжелой формой миотонической дистрофии могли сократить жизнь
- facioscapulohumeral MD — тип MD, который может развиться в детстве или в зрелом возрасте; прогрессирует медленно и обычно не опасно для жизни
- Becker MD — близкий родственник Duchenne MD, но развивается позже в детстве и протекает в меньшей степени; на продолжительность жизни обычно не так сильно влияет
- конечность-пояс MD — группа состояний, которые обычно развиваются в позднем детстве или в раннем взрослом возрасте; некоторые варианты могут быстро прогрессировать и быть опасными для жизни, тогда как другие развиваются медленно
- окулофарингеальный MD — тип MD, который обычно не развивается, пока человеку не исполнится от 50 до 60 лет, и не влияет на ожидаемую продолжительность жизни
- Emery-Dreifuss MD — тип MD, развивающийся в детстве или в раннем взрослом возрасте; большинство людей с этим заболеванием доживут как минимум до среднего возраста
Подробнее о видах МД и диагностировании МД.
Кто страдает мышечной дистрофией?
В Великобритании около 70 000 человек имеют MD или родственное заболевание.
Дюшенн MD является наиболее распространенным типом MD. В Великобритании ежегодно рождается около 100 мальчиков с диагнозом Дюшенна, а в Великобритании одновременно проживает около 2500 человек с этим заболеванием.
Миотонический MD является вторым по распространенности типом MD, которым страдает примерно 1 человек из 8000.
Считается, чтоFacioscapulohumeral MD поражает примерно 1 человека из 20 000 в Великобритании, что делает его третьим по распространенности MD.
Диагностика мышечной дистрофии
Для диагностики различных типов MD можно использовать множество различных методов. Возраст, в котором диагностируется состояние, будет варьироваться в зависимости от того, когда впервые начинают проявляться симптомы.
Диагностика включает некоторые или все из следующих этапов:
- исследование любых симптомов
- , обсуждая любую семейную историю MD
- медицинский осмотр
- анализы крови
- Электрические пробы нервов и мышц
- биопсия мышцы (при которой для исследования берут небольшой образец ткани)
Обратитесь к терапевту, если у вас или у вашего ребенка есть какие-либо симптомы MD.При необходимости ваш терапевт может направить вас в больницу для проведения дополнительных анализов.
Лечение мышечной дистрофии
Нет лекарства от MD, но ряд методов лечения может помочь с физическими недостатками и проблемами, которые могут развиться. Сюда могут входить:
- Помощь при передвижении, включая упражнения, физиотерапию и физические вспомогательные средства
- группы поддержки — чтобы справиться с практическим и эмоциональным воздействием MD
- хирургия — для исправления постуральных деформаций, таких как сколиоз
- лекарства, такие как стероиды для улучшения мышечной силы или ингибиторы АПФ и бета-блокаторы для лечения сердечных заболеваний
Новое исследование изучает способы исправления генетических мутаций и поврежденных мышц, связанных с MD.В настоящее время существуют многообещающие клинические испытания MD Duchenne.
Подробнее о лечении MD.
Информация о вас
Если у вас есть MD, ваша клиническая бригада передаст информацию о вас в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Узнайте больше о реестре.
Группы поддержки
MD может повлиять на вас как эмоционально, так и физически. Группы поддержки и организации могут помочь вам понять и смириться с вашим заболеванием. Они также могут предоставить полезные советы и поддержку тем, кто заботится о людях с МД.
Есть несколько национальных благотворительных организаций, которые предлагают поддержку людям, страдающим MD, например, Muscle Help Foundation и Muscular Dystrophy UK. Вы также можете спросить у своего терапевта или других лечащих вас медицинских работников о местных группах поддержки.
Есть также несколько национальных групп поддержки, которые продвигают исследования или оказывают поддержку конкретным типам MD, такие как Action Duchenne, Duchenne UK и Группа поддержки миотонической дистрофии.