Лечение болезни Штрюмпеля в Германии, клиника Вивантес
Неврологические патологии в современной медицине встречаются все чаще, а их специфика и особенности течения крайне разнообразны. Подобные заболевания способны поражать различные органы и системы организма, многие из них являются наследственными. К числу таких патологических процессов относится болезнь Штрюмпеля, прогрессирование которой затрагивает как структуры нервной системы, так и опорно-двигательного аппарата.
На базе неврологического отделения Вивантес в процессе диагностики и лечения болезни Штрюмпеля принимают участие одни из лучших врачей Германии. Специалисты нашей клиники проводят полный спектр исследований, используя для этого новейшее оборудование. Лечение осуществляется согласно инновационным методам консервативной терапии, при необходимости опытные хирурги проводят операции различной степени сложности.
Что такое болезнь Штрюмпеля
Болезнь Штрюмпеля, также известная как семейная спастическая параплегия – патологический процесс из группы дегенеративных миелопатий.
При данном заболевании затрагиваются пирамидные пути спинного мозга. В результате этого происходит поражение центральной нервной системы, последствием чего становятся нарушения со стороны опорно-двигательного аппарата. В подавляющем большинстве случаев болезнь Штрюмпеля затрагивает нижние конечности, механизм ее развития заключается в прогрессирующем парапарезе нижних конечностей. По мере развития заболевания нарастает гипертонус мышечных структур нижних конечностей.
Согласно статистике, манифестация заболевания приходится на возраст от 10 до 30 лет. Однако в обширном понимании дебют болезни Штрюмпеля возможен в любом возрасте от рождения и вплоть до 80 лет. Что касается причин возникновения данной патологии, речь идет о генетической предрасположенности, обусловленной генными мутациями.
Классификация патологического процесса тесно сопряжена с причинами его развития и включает такие виды спастической параплегии:
- аутосомно-доминантная – заболевание наблюдается у одного из родителей, риск его развития у ребенка составляет порядка 50%;
- аутосомно-рецессивная – оба родителя являются носителями дефектного гена (только носителями), риск возникновения патологии у ребенка – порядка 25%;
- Х-сцепленная – носителем дефектного гена выступают только женщины, однако, болезнь развивается у ребенка мужского пола.
Симптомы и диагностика болезни Штрюмпеля
Особенности клинической картины болезни во многом зависят от возраста, в котором началось ее развитие. Клинические признаки при этом затрагивают преимущественно нижние конечности, усугубляются по мере прогрессирования, вплоть до полной потери способности ходить.
К числу наиболее распространенных и характерных симптомов болезни Штрюмпеля специалисты клиники «Вивантес» относят:
- ощущение скованности в ногах;
- повышение мышечного тонуса нижних конечностей;
- шаткость походки;
- частые падения;
- гиперрефлексия обеих ног;
- снижение силы мышц нижних конечностей;
- на поздних стадиях прогрессирования возникает мышечная атрофия;
- при раннем дебюте заболевания отмечается хождение ребенка на цыпочках или отставание от сверстников в плане формирования навыков ходьбы.


В процессе диагностики особое внимание уделяется особенностям клинической картины, возрасту, когда заболевание дебютировало, данным общего осмотра и анамнеза семьи пациента.
Для постановки точного диагноза и разработки эффективной тактики лечения наши врачи выполняют ряд важных исследований:
- общеклинические анализы крови и мочи;
- биохимическое исследование крови;
- молекулярно-генетические исследования;
- магнитно-резонансная томография позвоночного столба в двух проекциях;
- электронейромиография;
- исследование вызванных потенциалов.
Также важная дифференциация болезни Штрюмпеля с рядом других патологий, в числе которых опухоли спинного мозга, боковой амиотрофический склероз, нейросифилис и другие.
Лечение болезни Штрюмпеля
Разработкой оптимального плана лечения в нашей клинике занимается целая команда квалифицированных врачей. Выбор методов зависит от целого ряда факторов, учитывая анамнез пациента, его возраст, степень прогрессирования заболевания, тяжесть симптомов.
Консервативное
На сегодняшний день основным направлением при болезни Штрюмпеля является консервативная терапия. При этом специфического лечения не существует, действия врачей сводятся к купированию симптомов и повышению качества жизни пациента.
Таким образом, основными применяемыми группами препаратов являются:
- миорелаксанты – снижают спастику и купируют мышечные спазмы;
- транквилизаторы – обладают схожим с миорелаксантами механизмом действия, способствуют расслаблению мышц;
- введение мышечные структуры ботулотоксина.
Улучшение состояния пациента также достигается благодаря индивидуально разработанным программам лечебной физкультуры, курсам массажа, физиопроцедурам. В некоторых случаях пациенту назначается ношение вспомогательных конструкций, например, ортезов.
Хирургическое
Оперативные вмешательства, направленные на лечение болезни Штрюмпеля проводятся лишь при наличии соответствующих показаний.
Реабилитация
Разрабатываемые реабилитационные программы направлены на закрепление успеха, достигнутого в рамках лечения. Все действия в данном случае сводятся к повышению качества жизни пациента и предотвращению утраты способности ходить. Для этого продолжаются курсы массажа, физиопроцедуры, занятия ЛФК, корректируются дозы препаратов.
Доктора
- Инсульт и сосудистые заболевания головного мозга
- Неврологическая реабилитация
- Рассеянный склероз
- Нейроиммунология
- Нейроинтенсивная терапия
- Эпилепсия
- Дифференциальная диагностика неэпилептических пароксизмов
- Длительный ЭЭГ — видеомониторинг
- Медицинская и немедицинская помощь при пароксизмах и осложнениях, связанных с эпилепсией
- Член Британской Медицинской Ассоциации
- Инструктор и член Европейской Академии по изучению Эпилепсии (EUREPA)
- Член Комиссии по психобиологии и Международной Лиги по борьбе с Эпилепсией (ILAE)
- Психоорганический синдром, Всемирная Федерация Обществ Биологической Психиатрии (WFSBP)
- Видеоконсультация
- Признанный международный эксперт в области болезни Паркинсона, дистонии и тремор
- Нейромускулярные заболевания
- Рассеянный склероз
- Лечение с использованием бутолотоксина (дистония, спастика)
- Лечение глубокой стимуляцией мозга
- Автор более 70 научных публикаций, явялется членом международных экспертных советов
- Всемирно признанный специалист в области инсульта, заболеваний периферической нервной системы и клинической электрофизиологии
- Автор свыше 40 оригинальных публикаций
- Член совета директоров Берлинского центра исследований инсульта (CSB)
- Гендерная медицина
- Психоонкология
- Биполярные аффективные расстройства
- Неврологическая реабилитация после инсульта, полученных черепно-мозговых травм и повреждений спинного мозга
- Реабилитационная терапия при болезни Паркинсона и дистонии
- Ботулинотерапия
- Лечение спастичности
- Неврологическая реабилитация с помощью интратекальной баклофеновой терапии и глубокой мозговой стимуляции
- Автор более 90 научных публикаций, член нескольких экспертных комиссий
Спастическая параплегия Штрюмпеля 4.
 73.18.3 GJC2 м.
73.18.3 GJC2 м.Исследуемый материал Цельная кровь (с ЭДТА)
Метод определенияСеквенирование
Выдаётся заключение врача-генетика!
Исследование мутаций в гене GJC2.
Болезнь Штрюмпеля относится к гетерогенным заболеваниям. Описано аутосомно-доминантное, аутосомно-рецессивное и Х-сцепленное наследование заболевания. В большинстве семей имеет место аутосомно-доминантное наследование болезни, соответствующее оригинальным описаниям Штрюмпеля. Именно для аутосомно-доминантной наследственной спастической параплегии обоснованно использование широко распространенного эпонимического термина болезнь Штрюмпеля. Наследственная спастическая параплегия характеризуется глиозным перерождением пирамидных путей в боковых и передних канатиках на грудном, поясничном уровне спинного мозга. На поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга. Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.
На поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга. Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.

Тип наследования.
Аутосомно- рецессивный.
Гены, ответственные за развитие заболевания.
Ген GJC2 белка щелевых контактов коннексин 47 (Сх47) (GAP JUNCTION PROTEIN, GAMMA-2). Ген расположен на хромосоме 1 в регионе 1q42.13. Содержит 2 экзона.
Мутации в этом гене приводят также к развитию гипомиелинизирующей лейкодистрофии тип 2, врождённой лимфедеме, тип 1С.
Патогенез и клиническая картина.
Наследственная спастическая параплегия характеризуется глиозным перерождением пирамидных путей в боковых и передних канатиках на грудном, поясничном уровне спинного мозга, на поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга.
Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.
Для болезни Штрюмпеля характерны следующие признаки проявления заболевания: постепенное развитие заболевания, скованность, быстрая утомляемость ног при ходьбе, стягивающие судороги в мышцах ног, спастическая походка, повышение сухожильных рефлексов, клонусы стоп и коленных чашечек, раннее появление сухожильных рефлексов, формирование контрактур и деформация стопы по типу «стопы Фридрейха». Верхние конечности вовлекаются редко и в поздней стадии. Чувствительность и интеллект не изменены. Иногда – поражение II (зрительный) и III (глазодвигательный) нервов, дизартрия, нистагм, атаксия, интенционный тремор.
Верхние конечности вовлекаются редко и в поздней стадии. Чувствительность и интеллект не изменены. Иногда – поражение II (зрительный) и III (глазодвигательный) нервов, дизартрия, нистагм, атаксия, интенционный тремор.
Характерным параклиническим признаком наследственной спастической параплегии является картина атрофии спинного мозга на всем протяжении (особенно в каудальных отделах), выявляемая при проведении МРТ.
Для данного типа заболевания характерно: позднее начало заболевания, медленно прогрессирующее течением, осложненное спастической параплегией, с нормальным или почти нормальным психомоторным развитием, нистагм отсутствует. Способность ходить больные сохраняют до зрелого возраста. Гетерозиготные члены семьи здоровы.
Частота встречаемости: заболевание редкое. Описана одна семья.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- GE Rudenskaya, AV Polyakov. Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.
- Orthmann-Murphy, J. L., Salsano, E., Abrams, C. K., Bizzi, A., Uziel, G., Freidin, M. M., Lamantea, E., Zeviani, M., Scherer, S. S., Pareyson, D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 132: 426-438, 2009.
- OMIM.
 Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.
Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.Спастическая параплегия Штрюмпеля, BSCL2 м.
Метод определения Секвенирование
Исследование мутаций в гене BSCL2.Тип наследования.
Аутосомно-доминантный.Гены, ответственные за развитие заболевания.
BSCL2.Ген расположен на хромосоме 11 в регионе 11q12.3. Содержит 11 экзонов.
Мутации в данном гене приводят также к развитию врожденной генерализованной липодистрофии тип 2; дистальной врождённой моторной нейропатии тип V.
Определение заболевания.
Семейная спастическая параплегия Штрюмпеля — это хроническое прогрессирующее наследственно-дегенеративное заболевание нервной системы, при котором поражаются с двух сторон пирамидные пути в боковых и передних канатиках спинного мозга. Ведущим клиническим синдромом является прогрессирующий нижний спастический парапарез. Болезнь Штрюмпеля относится к гетерогенным заболеваниям. Описаны случаи аутосомно-доминантного, аутосомно-рецессивного и Х-сцепленного наследования заболевания. В большинстве семей имеет место аутосомно-доминантное наследование болезни.Патогенез и клиническая картина.
Ген BSCL2 кодирует трансмембранный белок эндоплазматической сети сейпин. Наследственная спастическая параплегия характеризуется глиозным перерождением пирамидных путей в боковых и передних канатиках на грудном, поясничном уровне спинного мозга, на поздних стадиях болезни вовлекаются пирамидные волокна в стволе мозга и частичная гибель клеток Беца двигательной зоны коры полушарий большого мозга.
Первые симптомы могут развиваться в любом возрасте от 1-го до 7-го десятилетия жизни, что связано с генетической гетерогенностью.
Для болезни Штрюмпеля характерны следующие признаки проявления заболевания: постепенное развитие заболевания, скованность, быстрая утомляемость ног при ходьбе, стягивающие судороги в мышцах ног, спастическая походка, повышение сухожильных рефлексов, клонусы стоп и коленных чашечек, раннее появление сухожильных рефлексов, формирование контрактур и деформация стопы по типу «стопы Фридрейха». Верхние конечности вовлекаются редко и в поздней стадии. Чувствительность и интеллект не изменены. Иногда – поражение II (зрительный) и III (глазодвигательный) нервов, дизартрия, нистагм, атаксия, интенционный тремор.
Характерным параклиническим признаком наследственной спастической параплегии является картина атрофии спинного мозга на всем протяжении (особенно в каудальных отделах), выявляемая при проведении МРТ. Частота встречаемости: не установлена. Заболевание редкое.
Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- GE Rudenskaya, AV Polyakov. Hereditary spastic paraplegia in Russia: epidemiological, clinical and molecular aspects. TWS symposium on hereditary spastic paraplegias: www.hsp-info.de.
- Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Horl, G., Malli, R., Reed, J. A., Dierick, I., Verpoorten, N., Warner, T. T., Proukakis, C., Van den Bergh, P., Verellen, C., Van Maldergem, L., Merlini, L., De Jonghe, P., Timmerman, V., Crosby, A. H., Wagner, K. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nature Genet. 36: 271-276, 2004.
- OMIM.
Болезнь Штрюмпеля
Болезнь Штрюмпеля — генетически обусловленное заболевание, которое может быть как аутосомно-рецессивным, так и аутосомно-доминантным, характеризующееся поражением пирамидных путей и проявляющееся прогрессирующим повышением мышечного тонуса в нижних конечностях [3].
Впервые данное заболевание было описано в 1883 году. Описал его А. Strumpel. Он указал наличие наследственных форм параплегий, представив описания клинических случаев пациентов, для которых было характерно прогрессирование спастики и слабости в нижних конечностях. При этом отмечались негрубые нарушения вибрационной чувствительности и нарушения в работе мочевого пузыря.
Найти точные данные о распространенности данной патологии представляется затруднительным, они варьируют от 6 до 8 случаев на 100000 общей популяции.
Принято выделять две основных классификации: по типу наследования и по клиническому принципу [2].
По первой классификации данное заболевание делится на аутосомно-доминантные формы, аутосомно-рецессивные и Х-сцепленные. При постановке диагноза отмечается символ гена заболевания. Ссылаясь на англоязычные источники — SPG, что означает «spastic paraplegia gene» — ген спастической параплегии. Указывается цифра от 1 до 32 для обозначения хронологического порядка описания локуса [2].
По второй классификации выделяются простые и сложные формы заболевания. Простые — это те, при которых наследственные спастические параплегии являются основным симптомом, а сложные — сочетание с такими симптомами, как умственная отсталость, снижение остроты слуха, дегенерация сетчатки, атрофия дисков зрительных нервов, эпилепсия, церебральная атаксия, периферическая нейропатия. По статистике, сложные формы не превышают 10 % от общего числа заболеваний [2].
Патогенез Болезни Штрюмпеля изучен недостаточно. На данный момент выделяют четыре механизма развития заболевания, заключающихся в функционировании тех или иных генов. В эмбриогенезе нарушается развитие гена SPG1, в результате чего неправильно формируются аксоны нервных клеток в головном мозге, мозжечке, спинном мозге. Ошибка в гене L1CAM приводит к сбою в механизме дифференциации клеток и росте аксонов [1].
Изменения в гене SPG2 приводит к нарушению выработки миелина и созревания олигодендроцитов [1].
Таблица 1 | Локализация, тип наследования и продукты генов SPG
Однако вне зависимости от того, какое звено патогенеза участвует в развитии болезни Штрюмпеля, во всех описанных случаях происходит дегенерация аксонов, входящих в пирамидные пути и средние столбы спинного мозга [3].
Имеются описания уменьшения количества нейронов в пятом слое моторной коры и базальных ганглиях ГМ, мозжечке, переднем роге спинного мозга [3].
Несмотря на то что чаще всего заболевание проявляет себя на втором десятке жизни, бывают и бессимптомные носители генетических изменений. Для пациентов с ранним началом заболевания характерна тенденция к хождению на цыпочках. Те же, кого болезнь застает в более позднем возрасте, чаще всего жалуются на нарушения походки и частые падения, потерю равновесия при ходьбе, будто бы плохо ощущают опору стоп. Повышается мышечный тонус в ногах, на ранних стадиях заболевания спастичность отмечается только во время ходьбы, но с прогрессирование болезни Штрюмпеля она становится заметна и в покое. Мышечный тонус повышен в приводящих мышцах бедер, задней группе мышц бедра, камбаловидных мышцах [1,3].
Проявление мышечной слабости отмечается позже появления спастичности. Характерным симптомом является гиперрефлексия, возможны клонусы стоп.
Для некоторых форм заболевания характерны псевдобульбарные нарушения, характеризующиеся нарушениями функций мочевого пузыря. При этом стоит отметить, что чаще всего подобные проявления встречаются у пожилых женщин на далеко зашедших стадиях заболевания [3].
Динамика и скорость развития тех или иных симптомов заболевания сильно варьируют. Если пациент болен простой формой, прогрессирование медленное. Такие больные лишь без соответствующего лечения теряют способность к самостоятельному передвижению. При сложных формах к основным симптомом присоединяются и вторичные, как правило, со стороны нервной системы. В первую очередь проявляется деменция [2].
Диагностика болезни Штрюмпеля является проблемной. Необходимо обращать внимание на проявления заболевания и семейный анамнез. Ведущим симптомом является прогрессирующая спастическая параплегия [2].
Инструментальные методы исследований не всегда дают достаточно оснований для постановки диагноза. Использование метода МРТ спинного мозга позволяет выявить его дистрофию. Электронейромиография может использоваться как один из способов диагностики, однако она применяется редко и, в первую очередь, необходима для того, чтобы охарактеризовать сопутствующую нейропатию, если она, конечно же, есть. Определение соматосенсорных вызванных потенциалов нижних конечностей демонстрирует задержку проведения импульса по задним столбам СМ. Вызванные корковые потенциалы демонстрируют значительное снижение скорости проведения по кортико-спинальному тракту и снижение амплитуды вызванных потенциалов. Может применяться молекулярная диагностика, однако из-за своей дороговизны и малой доступности она практически не используется [1,2].
Электронейромиография может использоваться как один из способов диагностики, однако она применяется редко и, в первую очередь, необходима для того, чтобы охарактеризовать сопутствующую нейропатию, если она, конечно же, есть. Определение соматосенсорных вызванных потенциалов нижних конечностей демонстрирует задержку проведения импульса по задним столбам СМ. Вызванные корковые потенциалы демонстрируют значительное снижение скорости проведения по кортико-спинальному тракту и снижение амплитуды вызванных потенциалов. Может применяться молекулярная диагностика, однако из-за своей дороговизны и малой доступности она практически не используется [1,2].
Специфического лечения болезни Штрюмпеля не существует. Любое лечение направлено на устранение симптомов заболевания. Используются такие препараты, как баклофен, диазепам, дантролен. Применяются они как перорально, так и эндолюмбально при тяжелых формах спастичности. При назначениях любой формы препаратов придерживаются принципа «от меньшего к большему».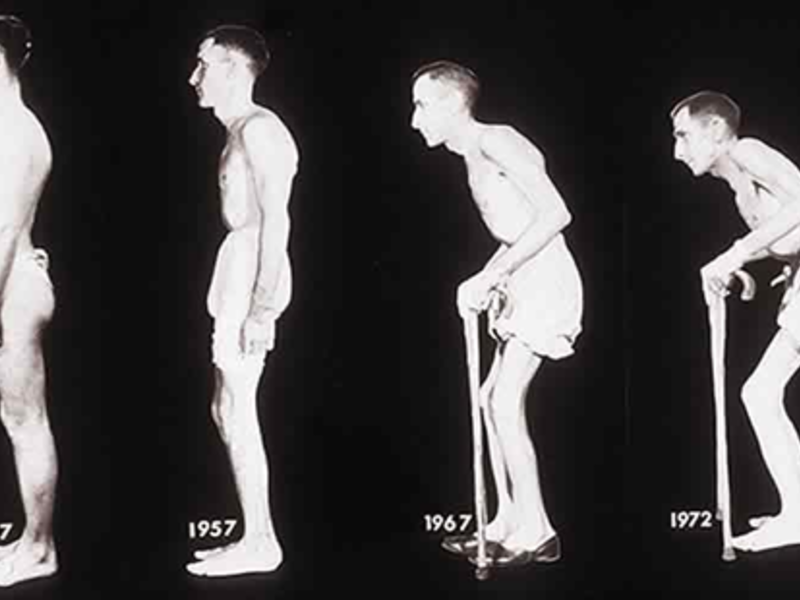 Начинают с минимальной дозы и при отсутствии эффекта ее увеличивают. Отмена препаратов производится постепенно, чтобы снизить вероятность развития синдрома отмены [1].
Начинают с минимальной дозы и при отсутствии эффекта ее увеличивают. Отмена препаратов производится постепенно, чтобы снизить вероятность развития синдрома отмены [1].
Таблица 2 | Принципы лекарственной терапии болезни Штрюмпеля
Также при лечении спастичности часто используется ботулотоксин. Он вводится в икроножные мышцы, заднюю группу мышц бедра. После его введения наблюдается улучшение подвижности. Также ботулотоксин является профилактической мерой укорочения мышечных сухожилий наряду с лечебной физкультурой [1].
Источники:
- Е. Д. Белоусова. Наследственные спастические параплегии. / E. Д. Белоусова // Российский вестник педиатрии — 2010 — С. 63–69.
- Е. И. Елагина. Наследственно-дегенеративные заболевания нервной системы. Болезнь Штрюмпеля или семейная спастическая параплегия./ Е. И. Елагина, Е. Ю. Курыцина // Проблемы медицины в современных условия — 2014 — С. 205–209
- Г. Е. Руденская. Распространенные форма наследственных спастических параплегий. / Г .Е. Руденская, И. Г. Сермягина, С. Н. Иллариошкин // Журнал неврологии и и психиатрии им. С.С. Корсакова — 2019 — С. 94–104
 / Г .Е. Руденская, И. Г. Сермягина, С. Н. Иллариошкин // Журнал неврологии и и психиатрии им. С.С. Корсакова — 2019 — С. 94–104
/ Г .Е. Руденская, И. Г. Сермягина, С. Н. Иллариошкин // Журнал неврологии и и психиатрии им. С.С. Корсакова — 2019 — С. 94–104Болезнь Штрюмпеля — что это, как лечить
Болезнь Штрюмпеля – это патология, при которой у человека наблюдается сильная слабость в области ног. Происходит подобное из-за повышения мышечного тонуса в нижних конечностях. Болезнь имеет наследственный характер, при этом она способна начать прогрессировать в любом возрасте.
При спастической параплегии (другое наименование болезни Штрюмпеля) наблюдается негативное влияние на работу центральной нервной системы. При этом наиболее часто патология возникает в возрасте от 10 до 30 лет.
Её главным симптомом является паралич нижних конечностей. При возникновении подозрениях на болезнь Штрюмпеля человеку следует в обязательном порядке обратиться в больницу и пройти диагностику патологии.
Провоцирующие факторы и классификация патологии
Учёным удалось доказать, что болезнь Штрюмпеля имеет наследственный характер. По этой причине при появлении отклонения у ребёнка будет наблюдаться такая же патология хотя бы у одного из родителей. Если мама или папа являются носителем гена, тогда примерно в 50% случаях он передаётся детям. Когда оба родителя страдают от болезни Штрюмпеля, тогда вероятность появления паралича – 25%. Также существует X-сцепленный тип передачи гена. В этом случае женщины являются носителями патологии, но страдают от неё исключительно мужчины.
По этой причине при появлении отклонения у ребёнка будет наблюдаться такая же патология хотя бы у одного из родителей. Если мама или папа являются носителем гена, тогда примерно в 50% случаях он передаётся детям. Когда оба родителя страдают от болезни Штрюмпеля, тогда вероятность появления паралича – 25%. Также существует X-сцепленный тип передачи гена. В этом случае женщины являются носителями патологии, но страдают от неё исключительно мужчины.
- Болезнь появляется из-за того, что происходит мутация в 19 хромосоме в гене ретикулон.
- При этом стоит отметить, что болезнь бывает разных типов. Врачи её классифицируют в зависимости от тяжести клинических симптомов.
Выделяют следующие виды:
- Неосложнённая форма. В этом случае имеется спастический нижний парапарез. Другие отклонения в состоянии здоровья отсутствуют. В этой ситуации человек может поддерживать нормальное качество жизни, потому как он страдает только от нарушения двигательной активности нижних конечностей.

- Осложнённый вид. Кроме нижнего парапареза будет наблюдаться атрофия зрительного нерва, нарушение речи, а также мозжечковая атаксия. Человек нередко страдает от порока сердца, проблем со слухом, а также эпилепсии. В некоторых случаях может присутствовать задержка психического развития, а также деформация стоп.
Также патологию принято разделять в зависимости от причины появления болезни Штрюмпеля:
- Идиопатический вид. Он диагностируется в том случае, если у членов семьи не была выявлена патология. В такой ситуации провоцирующий фактор остаётся неизвестным.
- Семейный тип. Болезнь передаётся от родителей к детям. При этом не во всех случаях потомки обязательно страдают от паралича нижних конечностей. Болезнь Штрюмпеля может перескочить через поколение, а также передаваться исключительно представителям мужского пола.
Дополнительно патологию классифицируют в зависимости от того, в каком возрасте она возникла. Отклонение может появиться, когда человеку ещё не исполнилось 35 лет. Преимущественно наблюдается спастика нижних конечностей, реже присутствует слабость. Хождение затруднено, но при этом человек не утрачивает полностью двигательную функцию.
Отклонение может появиться, когда человеку ещё не исполнилось 35 лет. Преимущественно наблюдается спастика нижних конечностей, реже присутствует слабость. Хождение затруднено, но при этом человек не утрачивает полностью двигательную функцию.
У некоторых людей патология появляется после 35 лет. В такой ситуации присутствует значительная мышечная слабость, нарушается функция мочевыводящих путей, сама болезнь развивается значительно быстрее, чем в случае возникновения патологии до 35 лет.
Только при обращении к врачу можно подтвердить наличие болезни Штрюмпеля. По этой причине сразу после появления первых симптомов необходимо посетить медицинского специалиста. В этой ситуации можно будет однозначно понять, с чем именно приходится иметь дело.
Также врач сможет назначить схему лечения, которая будет эффективна в конкретной ситуации.
Клиническая картина
Семейная спастическая параплегия развивается постепенно, поэтому поначалу симптомы не проявляются ярко. Если патология начинает развиваться в детском возрасте, тогда у ребёнка будут наблюдаться трудности при обучении ходьбе.
Если патология начинает развиваться в детском возрасте, тогда у ребёнка будут наблюдаться трудности при обучении ходьбе.
Нередко несовершеннолетний может в основном ходить на цыпочках, при этом будет редко полностью наступать на ступни. В этом случае людям рекомендуется проконсультироваться с медицинским специалистом по поводу того, может ли у ребёнка быть болезнь Штрюмпеля.
При её наличии важно будет сразу начать лечение, чтобы была возможность улучшить состояние человека.
Если патология возникает во взрослом возрасте, то сначала человек начнёт испытывать трудности при ходьбе. Ноги будут значительно быстрее утомляться, чем раньше. Могут возникать судороги, а также проблемы со сгибанием конечностей в коленях и тазобедренном суставе. Нередко пациент может часто падать из-за того, что нарушается функция конечностей.
Постепенно симптомы начинают развиваться и становиться интенсивнее. Из-за этого человеку тяжелее передвигаться, непосредственно трудно двигать стопами.
Мышечная слабость появляется значительно позже, потому как должно пройти время после возникновения заболевания.
Если наблюдается рецессивная форма болезни Штрюмпеля, тогда уменьшение мышечной силы может прогрессировать в течение нескольких лет. При доминантных формах патологии данный период наступает позже.
Мышечный тонус повышается с самого начала после появления патологии. Мускулатура не может до конца расслабиться, из-за чего она всё время напряжена.
Спастика наблюдается в глубокой области трехглавой мышцы голени, а также в бедренных мышцах. Спастичность может быть несимметричной, при этом она присутствует только в одной конечности.
Во время врачебного осмотра выявляется повышение тонуса сразу с двух сторон. Присутствует гиперрефлексия обеих нижних конечностей.
На поздних стадиях болезни Штрюмпеля появляются атрофические изменения в мышцах нижних конечностей. Они вызваны тем, что частично или полностью снизилась двигательная активность из-за пареза.
Они вызваны тем, что частично или полностью снизилась двигательная активность из-за пареза.
В некоторых ситуациях может наблюдаться атрофия мышц верхних конечностей. Также может начаться нарушение работы органов малого таза, из-за чего человек страдает от недержания мочи.
Данный симптом наиболее характерен для пожилых пациентов, при этом он может наблюдаться и у молодых людей.
При сложных видах болезни Штрюмпеля у человека можно наблюдать проявления неврологии:
- Когнитивные нарушения. У пациента значительно снижаются интеллектуальные способности, наблюдается деменция и олигофрения.
- Может начаться эпилепсия на фоне болезни Штрюмпеля. При этом приступы могут быть как незначительными, так и глобальными.
- Дизартрия. В этом случае пациент имеет проблемы при выражении своих мыслей, ему бывает трудно понимать речь других людей. Он может забывать то, что недавно услышал. Также нередко отсутствует возможность быстро и правильно подбирать слова для выражения своих мыслей.

- Нарушение слуха. В этом случае у человека значительно снижается слуховая функция. При этом данное изменение будет постепенно прогрессировать.
Конкретные симптомы зависят от состояния пациента, возраста, а также от формы семейной спастической параплегии.
В любом случае при возникновении первых признаков болезни важно незамедлительно обратиться к врачу для того, чтобы пройти диагностику. Только после полного обследования удастся подтвердить свой диагноз.
Также крайне важно определить конкретную форму патологии для того, чтобы можно было подобрать наиболее правильное лечение.
Методы диагностики
При диагностике болезни Штрюмпеля используются разные методики. Пациента направляют к неврологу, если имеется осложнённая форма патологии.
- Потребуется убедиться, что у человека отсутствует опухоль спинного мозга, рассеянный склероз, нейросифилис, а также сосудистая миелопатия. Также потребуется понять, есть ли у пациента лейкодистрофия, с этой целью проводится МРТ головного мозга. Данное обследование позволяет визуализировать орган и оценить его состояние с разных ракурсов. Благодаря этому удаётся точно понять, имеются ли в черепно-мозговой коробке какие-либо поражения. В некоторых случаях могут быть выявлены атрофические изменения в коре головного мозга.
- Достаточно часто выполняется МРТ позвоночника, это нужно для визуализации дегенеративно-атрофических процессов в передних и боковых столбах. Поражение может находиться на уровне грудных или поясничных элементов спинного мозга.
 Данное обследование позволяет визуализировать орган и оценить его состояние с разных ракурсов. Благодаря этому удаётся точно понять, имеются ли в черепно-мозговой коробке какие-либо поражения. В некоторых случаях могут быть выявлены атрофические изменения в коре головного мозга.
Данное обследование позволяет визуализировать орган и оценить его состояние с разных ракурсов. Благодаря этому удаётся точно понять, имеются ли в черепно-мозговой коробке какие-либо поражения. В некоторых случаях могут быть выявлены атрофические изменения в коре головного мозга.- Дополнительным методом, который применяется для диагностики болезни Штрюмпеля, выступает электронейромиография. Данное исследование помогает определить наличие нейропатии, а также её степень. Потребуется обследовать соматосенсорные ВП, чтобы убедиться в наличии задержки проведения по задним спинномозговым столбам. Также выполняется исследование корковых ВП, при этом будет наблюдаться снижение скорости проведения по корково-спинальному пути.
- В обязательном порядке медицинский специалист узнает, есть ли у кого-то в семье болезнь Штрюмпеля. В большинстве случаев она имеется у ближних родственников. В такой ситуации подтверждается наследственный фактор, который привёл к возникновению заболевания. Бывает и такое, что никто из близких родственников не страдал от патологии. В такой ситуации точная причина, из-за которой появилась болезнь Штрюмпеля, остаётся невыясненной.

- После всех обследований можно будет сделать однозначный вывод по поводу того, с чем именно приходится иметь дело. Врач сможет назначить подходящее лечение, с помощью которого удастся значительно улучшить состояние пациента. Важно предотвратить развитие патологии, а также улучшить двигательную активность. Если своевременно начать терапию и следовать всем рекомендациям врача, тогда можно будет значительно быстрее нормализовать своё самочувствие.
Методы лечения
- На текущий момент нет конкретного способа, который мог бы точно предотвратить развитие болезни Штрюмпеля или вовсе от неё избавить человека. Есть разные методики, которые позволят ослабить проявления патологии, улучшить физическое и эмоциональное состояние, а также незначительно замедлить ход недуга.
- Врачи прописывают пациентам миорелаксанты, которые требуются для нормализации мышечного тонуса. Благодаря данным таблеткам удаётся расслабить мускулатуру, что положительно сказывается на самочувствии пациента. Среди популярных препаратов можно отметить Мидокалм, Баклофен и Скутамил. Также нередко применяются транквилизаторы, такие как Седуксен и Тазепам. С их помощью удаётся улучшить психоэмоциональное состояние человека. Для поддержания организма рекомендуется использовать витамины группы B.
 Есть разные методики, которые позволят ослабить проявления патологии, улучшить физическое и эмоциональное состояние, а также незначительно замедлить ход недуга.
Есть разные методики, которые позволят ослабить проявления патологии, улучшить физическое и эмоциональное состояние, а также незначительно замедлить ход недуга.- Поначалу средства при семейной спастической параплегии назначаются в минимальной дозировке, при этом постепенно доза увеличивается. Когда уменьшается спастика, приём препарата врач перестаёт корректировать. При появлении побочных эффектов наращивание дозы сразу прекращается. Вполне возможно, что её потребуется снизить, чтобы избавиться от негативного действия лекарства. Если таблетки окажутся неэффективными в конкретной ситуации, тогда человеку назначат инъекции.
- При тяжёлых формах болезни будет прописана постоянная инфузия Баклофена. Его потребуется вводить в цереброспинальную жидкость. Помимо этого, применяется Ботулотоксин, его вкалывают в задние бедренные мышцы, а также в икры.
 Вполне возможно, что её потребуется снизить, чтобы избавиться от негативного действия лекарства. Если таблетки окажутся неэффективными в конкретной ситуации, тогда человеку назначат инъекции.
Вполне возможно, что её потребуется снизить, чтобы избавиться от негативного действия лекарства. Если таблетки окажутся неэффективными в конкретной ситуации, тогда человеку назначат инъекции.- Кроме медикаментозного лечения человеку рекомендуется делать специальную гимнастику, а также ходить на физиопроцедуры. Нередко пациенту требуется записаться на массаж, а также принимать целебные ванны. Все пациенты должны постоянно наблюдаться у ортопеда. Он должен следить за тем, насколько помогает лечение в конкретной ситуации. При необходимости медицинский специалист будет корректировать схему терапии.
- В редких ситуациях человеку может потребоваться носить протезы. Также некоторых пациентов направляют на оперативное вмешательство для лечения пассивных ограничений в области суставов.

Следует понимать, что болезнь Штрюмпеля не угрожает жизни человека, при этом качество существования снижается. Может значительно нарушиться трудоспособность пациента. Патология имеет свойство прогрессировать, из-за чего постепенно состояние гражданина ухудшается. В большинстве случаев у пациентов сохраняется способность самостоятельно передвигаться, но при этом существует утрата двигательной функции. Патология не влияет на продолжительность жизни, если болезнь Штрюмпеля не осложнена иными отклонениями. Болезнь Штрюмпеля: симптомы и причины появления патологии Ссылка на основную публикацию
Болезнь Штрюмпеля
Болезнь Штрюмпеля — генетически обусловленное заболевание, которое может быть как аутосомно-рецессивным, так и аутосомно-доминантным, характеризующееся поражением пирамидных путей и проявляющееся прогрессирующим повышением мышечного тонуса в нижних конечностях [3].
Впервые данное заболевание было описано в 1883 году. Описал его А. Strumpel. Он указал наличие наследственных форм параплегий, представив описания клинических случаев пациентов, для которых было характерно прогрессирование спастики и слабости в нижних конечностях. При этом отмечались негрубые нарушения вибрационной чувствительности и нарушения в работе мочевого пузыря.
Он указал наличие наследственных форм параплегий, представив описания клинических случаев пациентов, для которых было характерно прогрессирование спастики и слабости в нижних конечностях. При этом отмечались негрубые нарушения вибрационной чувствительности и нарушения в работе мочевого пузыря.
Найти точные данные о распространенности данной патологии представляется затруднительным, они варьируют от 6 до 8 случаев на 100000 общей популяции.
Принято выделять две основных классификации: по типу наследования и по клиническому принципу [2].
По первой классификации данное заболевание делится на аутосомно-доминантные формы, аутосомно-рецессивные и Х-сцепленные. При постановке диагноза отмечается символ гена заболевания. Ссылаясь на англоязычные источники — SPG, что означает «spastic paraplegia gene» — ген спастической параплегии. Указывается цифра от 1 до 32 для обозначения хронологического порядка описания локуса [2].
По второй классификации выделяются простые и сложные формы заболевания.
Простые — это те, при которых наследственные спастические параплегии являются основным симптомом, а сложные — сочетание с такими симптомами, как умственная отсталость, снижение остроты слуха, дегенерация сетчатки, атрофия дисков зрительных нервов, эпилепсия, церебральная атаксия, периферическая нейропатия. По статистике, сложные формы не превышают 10 % от общего числа заболеваний [2].
Патогенез Болезни Штрюмпеля изучен недостаточно. На данный момент выделяют четыре механизма развития заболевания, заключающихся в функционировании тех или иных генов.
В эмбриогенезе нарушается развитие гена SPG1, в результате чего неправильно формируются аксоны нервных клеток в головном мозге, мозжечке, спинном мозге.
Ошибка в гене L1CAM приводит к сбою в механизме дифференциации клеток и росте аксонов [1].
Изменения в гене SPG2 приводит к нарушению выработки миелина и созревания олигодендроцитов [1].
Таблица 1 | Локализация, тип наследования и продукты генов SPG
Однако вне зависимости от того, какое звено патогенеза участвует в развитии болезни Штрюмпеля, во всех описанных случаях происходит дегенерация аксонов, входящих в пирамидные пути и средние столбы спинного мозга [3].![]()
Имеются описания уменьшения количества нейронов в пятом слое моторной коры и базальных ганглиях ГМ, мозжечке, переднем роге спинного мозга [3].
Несмотря на то что чаще всего заболевание проявляет себя на втором десятке жизни, бывают и бессимптомные носители генетических изменений. Для пациентов с ранним началом заболевания характерна тенденция к хождению на цыпочках.
Те же, кого болезнь застает в более позднем возрасте, чаще всего жалуются на нарушения походки и частые падения, потерю равновесия при ходьбе, будто бы плохо ощущают опору стоп.
Повышается мышечный тонус в ногах, на ранних стадиях заболевания спастичность отмечается только во время ходьбы, но с прогрессирование болезни Штрюмпеля она становится заметна и в покое. Мышечный тонус повышен в приводящих мышцах бедер, задней группе мышц бедра, камбаловидных мышцах [1,3].
Проявление мышечной слабости отмечается позже появления спастичности. Характерным симптомом является гиперрефлексия, возможны клонусы стоп.
Для некоторых форм заболевания характерны псевдобульбарные нарушения, характеризующиеся нарушениями функций мочевого пузыря. При этом стоит отметить, что чаще всего подобные проявления встречаются у пожилых женщин на далеко зашедших стадиях заболевания [3].
Динамика и скорость развития тех или иных симптомов заболевания сильно варьируют. Если пациент болен простой формой, прогрессирование медленное.
Такие больные лишь без соответствующего лечения теряют способность к самостоятельному передвижению.
При сложных формах к основным симптомом присоединяются и вторичные, как правило, со стороны нервной системы. В первую очередь проявляется деменция [2].
Диагностика болезни Штрюмпеля является проблемной. Необходимо обращать внимание на проявления заболевания и семейный анамнез. Ведущим симптомом является прогрессирующая спастическая параплегия [2].
Инструментальные методы исследований не всегда дают достаточно оснований для постановки диагноза. Использование метода МРТ спинного мозга позволяет выявить его дистрофию.
Использование метода МРТ спинного мозга позволяет выявить его дистрофию.
Электронейромиография может использоваться как один из способов диагностики, однако она применяется редко и, в первую очередь, необходима для того, чтобы охарактеризовать сопутствующую нейропатию, если она, конечно же, есть.
Определение соматосенсорных вызванных потенциалов нижних конечностей демонстрирует задержку проведения импульса по задним столбам СМ.
Вызванные корковые потенциалы демонстрируют значительное снижение скорости проведения по кортико-спинальному тракту и снижение амплитуды вызванных потенциалов. Может применяться молекулярная диагностика, однако из-за своей дороговизны и малой доступности она практически не используется [1,2].
Специфического лечения болезни Штрюмпеля не существует. Любое лечение направлено на устранение симптомов заболевания. Используются такие препараты, как баклофен, диазепам, дантролен.
Применяются они как перорально, так и эндолюмбально при тяжелых формах спастичности.
 При назначениях любой формы препаратов придерживаются принципа «от меньшего к большему». Начинают с минимальной дозы и при отсутствии эффекта ее увеличивают.
При назначениях любой формы препаратов придерживаются принципа «от меньшего к большему». Начинают с минимальной дозы и при отсутствии эффекта ее увеличивают.Отмена препаратов производится постепенно, чтобы снизить вероятность развития синдрома отмены [1].
Таблица 2 | Принципы лекарственной терапии болезни Штрюмпеля
Также при лечении спастичности часто используется ботулотоксин. Он вводится в икроножные мышцы, заднюю группу мышц бедра. После его введения наблюдается улучшение подвижности. Также ботулотоксин является профилактической мерой укорочения мышечных сухожилий наряду с лечебной физкультурой [1].
Источники:
- Е. Д. Белоусова. Наследственные спастические параплегии. / E. Д. Белоусова // Российский вестник педиатрии — 2010 — С. 63–69.
- Е. И. Елагина. Наследственно-дегенеративные заболевания нервной системы. Болезнь Штрюмпеля или семейная спастическая параплегия./ Е. И. Елагина, Е. Ю. Курыцина // Проблемы медицины в современных условия — 2014 — С. 205–209
- Г. Е. Руденская. Распространенные форма наследственных спастических параплегий./ Г .Е. Руденская, И. Г. Сермягина, С. Н. Иллариошкин // Журнал неврологии и и психиатрии им. С.С. Корсакова — 2019 — С. 94–104
 Ю. Курыцина // Проблемы медицины в современных условия — 2014 — С. 205–209
Ю. Курыцина // Проблемы медицины в современных условия — 2014 — С. 205–209Симптомы и лечение болезни Штрюмпеля
В последние годы наблюдается неблагоприятная тенденция к появлению в популяции наследственных заболеваний. Большинство из них диагностируется в детском или юношеском возрасте, но для болезни Штрюмпеля может быть характерен и более поздний дебют.
Согласно данным статистики, в среднем эта патология составляет около 4,1 случая на 100 тысяч населения. Заподозрить развитие недуга довольно сложно, так как приходиться проводить дифференциальный диагноз с другими заболеваниями, которые имеют такие же симптомы.
Если еще около 60-ти лет назад наличие такой параплегии было приговором, то современные средства реабилитации позволяют вывести жизнь пациента на более комфортный уровень.
Спастическая семейная параплегия (она же болезнь Штрюмпеля) — это заболевание наследственного характера, которое сопровождается поражением передних и боковых спинномозговых столбов преимущественно на уровне поясницы.
В качестве основного проявления недуга выступает нарушение подвижности нижних конечностей. Впервые об этом заболевании заговорили еще в XIX веке, а в 1883 году немецкий ученый-клиницист Штрюмпель впервые описал эту патологию.
В зависимости от характера клинических проявлений различают осложненные и неосложненные формы.
Основным субстратом болезни выступают неуклонно прогрессирующие глиозные (мелкоочаговые поражения нервной ткани) изменения пирамидных путей в боковых и передних столбах спинного мозга.
Также могут отмечаться процессы атрофии в передних рогах спинного мозга, нарушение целостности мозжечковых проводящих путей, уменьшение числа действующих нейронов.
Симптомы могут манифестировать в любом возрасте: от 10-ти до 70-ти лет жизни пациента.
Спастическая семейная параплегия может наследоваться несколькими способами. При аутосомно-доминантном типе один из родителей имеет доминантный ген, который в 50% случаев наследует потомство.
При аутосомно-рецессивном типе, когда оба родителя являются носителями аномального гена, эта вероятность составляет 25%.
Если же в основе патологии лежит X-сцепленное наследование, то болеют только представители сильного пола, а женщины вступают в качестве носительниц.
Если болезнь манифестирует в юном возрасте, среди первых симптомов наблюдается задержка физического развития. Ребенок позже начинает стоять и ходить, а также кажется довольно неуклюжим. Дебют болезни в более взрослом возрасте сопровождается возникновением затруднений при беге, ходьбе, частыми спотыканиями и падениями.
Пациенты жалуются на скованность ног, невозможность полностью опереться на стопу, повышение мышечного тонуса. В период первого появления параплегии повышение тонуса мышц носит перемежающийся характер: усиливается при активной деятельности и пропадает в покое. Спазмы преимущественно локализованы в мышечных группах бедра и голени.
Спазмы преимущественно локализованы в мышечных группах бедра и голени.
Нередко больной жалуется на поражение только одной ноги, но при объективном осмотре выявляется патология обеих конечностей.
Для заболевания характерно медленное и постепенное развитие: реабилитация и лечебные мероприятия замедляют этот процесс. При аутосомно-рецессивной форме недуга парез (мышечная слабость) появляется через несколько лет, при аутосомно-доминантном типе это может занять чуть больше времени. Характерно нарушение чувствительности (полиневропатия) при осложненном заболевании.
Поздние стадии болезни характеризуются развитием атрофических изменений мускулатуры нижних конечностей. Они возникают на фоне обездвиженности при выраженном парезе.
При прогрессировании недуга в патологический процесс могут вовлекаться мышцы верхних конечностей и малого таза (это приводит к недержанию мочи).
Среди осложненных форм заболевания преобладает неврологическая симптоматика: снижение когнитивных функций, эпилептические припадки, псевдобульбарный синдром.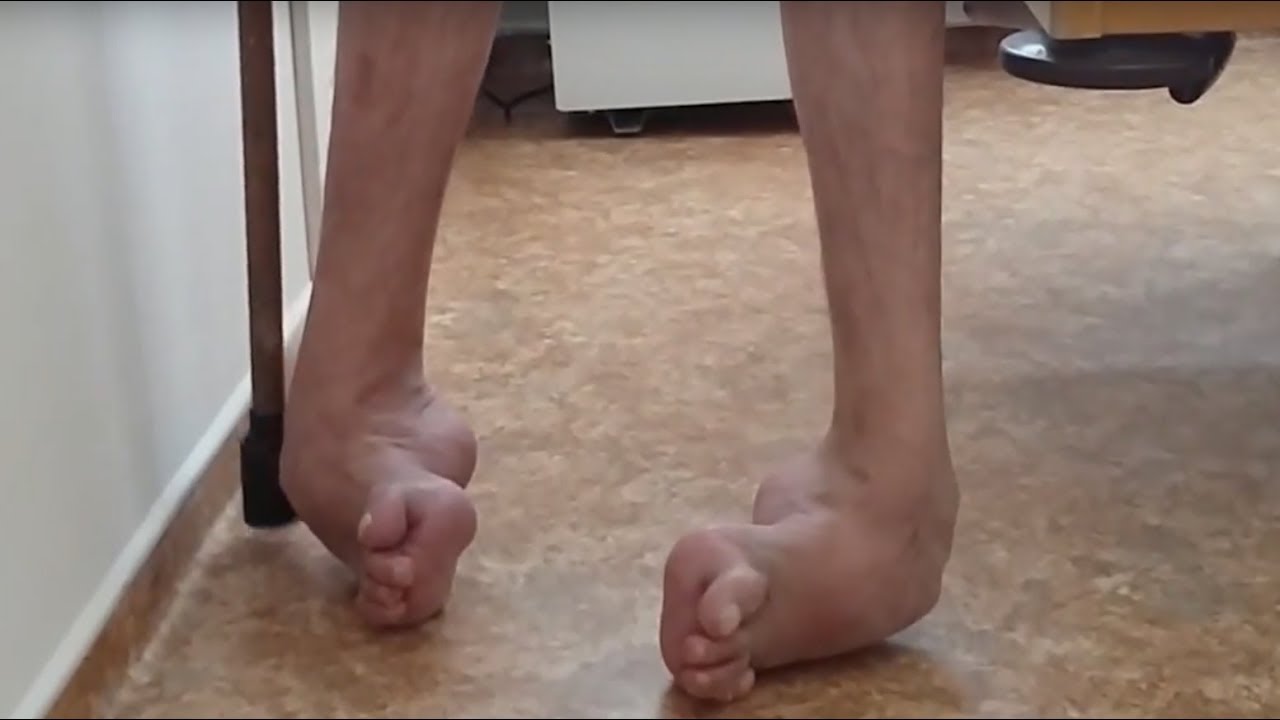 В отдельных случаях наблюдается снижение остроты зрения и слуха.
В отдельных случаях наблюдается снижение остроты зрения и слуха.
Болезнь Штрюмпеля: причины, симптомы и лечение параплегии
Болезнь Штрюмпеля – это болезнь, при которой в ногах начинает появляться слабость. Происходит это из-за повышенного мышечного тонуса в конечностях. Заболевание считается наследственным и может начать развиваться в любом возрасте.
Онлайн консультация по заболеванию «Болезнь Штрюмпеля».
Задайте бесплатно вопрос специалистам: Невролог.
Спастическая параплегия (другое название болезни Штрюмпеля) изучается уже больше века, однако, до сих пор исследования не завершились. Удалось выяснить, что патология оказывает влияние на работу центральной нервной системы и головного мозга. К тому же она медленно, но все же прогрессирует.
Наследственная проявившаяся спастическая параплегия передается по нескольким типам:
- Аутосомно-доминантный. Один из родителей ‒ носитель, тогда в 55% и даже в 100% случаев заболевание передается детям.
- Аутосомно-рецессивный. Двоим родителям поставлен диагноз носители заболевания, тогда риск развития патологии у детей составляет 25-50%.
- Х-сцепленный. Для этого типа характерно то, что носителями являются женщины, а заболевание развивается у мужчин.

Недавно зарубежные ученые обнаружили ген, вызывающий данный патологический процесс.
В медицине такая болезнь делится на несколько форм, которые имеют специфические симптомы:
- несложная форма, для которой характерен паралич ног, при этом иные патологии отсутствуют;
- осложненная форма – кроме паралича ног, наблюдается атрофия зрительного нерва, эпилептические приступы, ухудшение слуха, патологии при развитии сердца, а также неправильное развитие ступней.
По генетическим признакам наследственная развивающаяся спастическая параплегия делится на:
- семейную – передача заболевания происходит от поколения к поколению;
- идиопатическую – в истории болезни семьи эта патология не зафиксирована.

Также патология делится на типы, которые различны для каждого возраста:
- Первый тип. Характерен для людей до тридцати пяти лет, при этом спастическая параплегия ног проявляется чаще, чем слабость и трудности при хождении.
- Второй тип. Патология развивается после тридцати пяти лет. Проявляется слабость в мышцах, болезнь развивается немного быстрее.
Также есть классификация болезни по генному типу – на сегодняшний день тип SPG 4 самый распространенный. Треть пациентов страдает именно от него.
Болезнь Штрюмпеля проявляет симптомы довольно медленно:
- Основной признак в раннем детстве, когда болезнь только начинает развиваться, – дети трудно учатся ходить, плохо формируется этот навык.
- У подростков симптомы этой патологии таковы: трудности при ходьбе, быстрая утомляемость нижних конечностей, частые судороги, проблемы при сгибании ног в коленных суставах и тазобедренных, человек часто падает.
- Позже признаки становятся ярче, все движения затруднительны, больным тяжело двигать стопами.
- Проявление мышечной слабости наблюдается спустя длительное время после того, как начала развиваться спастическая параплегия.
- Для рецессивной формы такой период затянется на несколько лет. При доминантной форме период затягивается на более долгий срок.
- Тонус мышц начинает повышаться с самого детства, в результате чего они расслабиться не могут.
- Только одна нижняя конечность может иметь нарушения в движениях, однако, при обследовании тонус мышц фиксируется в обеих ногах.
- На фоне гиперрефлексии наблюдаются патологические рефлексы стоп разгибательного и сгибательного характера, а также синкинезия.
- На поздних этапах болезни, мышцы ног начинают атрофироваться, происходит полное либо частичное снижение их активности.
- При патологиях SPG 10 и SPG 17, отмирание мышечных волокон наблюдается в руках. На поздних этапах появляются сбои в работе органов таза и непроизвольное выделение мочи.
- Если диагностирована форма SPG 19, тогда вытекание мочи начинается с самого начала заболевания, но если это другая форма, то недержание больше характерно для больных преклонного возраста.
В сложных случаях появляются другие симптомы, а именно:
- нарушение интеллектуальных способностей;
- приступы эпилепсии;
- ретинопатия;
- ухудшение слуха и многие другие.
Необходимо отметить, что данный патологический процесс проявляет признаки не внезапно, а постепенно. Поэтому больной долгое время не идет в больницу. Но если обращение было своевременным, тогда прогноз лечения патологии будет благоприятным.
- Диагностика болезни Штрюмпеля подразумевает: визуальный осмотр врача, аппаратное обследование и лабораторные исследования.
- Первое, что уточняет врач во время осмотра, есть ли подобные жалобы у старших членов семьи.
- Кроме этого, необходимо:
- Опросить больного, чтобы выяснить, как давно появились подобные признаки, а именно: трудности при разговоре, слабость в ногах и другие.
- Определить патологические рефлексы и мышечный тонус, поскольку болезнь Штрюмпеля характеризуется снижением тонуса при повторном движении.
- Назначить компьютерную, а также магнитно-резонансную томографию. С их помощью исследуют спинной мозг, выявляют любые нарушения в нем. К сожалению, вероятность выявления изменений небольшая.
- Провести генетический анализ, который можно сделать еще в период внутриутробного развития.

Кроме описанных способов диагностики, проводится и дифференциальное обследование. Это поможет врачу исключить другие патологии, такие как нейросифилис, опухоли спинного мозга и другие, а МРТ поможет отличить это заболевание от лейкодистрофии.
Аппарат МРТ
Лечение болезни Штрюмпеля паллиативное. На сегодняшний день нет эффективных методов терапии, способных вылечить семейный спастический диагностированный паралич Штрюмпеля. Как и нет методов, чтобы остановить или замедлить его развитие.
Терапия поможет лишь снизить интенсивность проявления симптомов, а также улучшить психоэмоциональное и физическое состояние.
Врач назначает следующие группы препаратов:
- миорелаксанты;
- транквилизаторы;
- витамины группы В.

Прием этих лекарственных средств должен начинаться с минимальной дозировки, позже ее необходимо увеличивать. Как только спастика немного снизится, увеличение дозировки прекращается.
Если на фоне увеличения дозы появляются побочные явления, тогда ее необходимо начать снижать. Если прием медикаментозных средств невозможен или же они не дают ожидаемого результата, врач назначает инъекции.
В запущенных ситуациях лекарство вводят в цереброспинальную жидкость.
Кроме того, используют и другие методы терапии, а именно:
- лечебная гимнастика;
- физиотерапия (лечебный массаж, ванны и парафинолечение).
Все пациенты с диагнозом болезнь Штрюмпеля, а также те, кто прошел лечение, состоят на учете у ортопеда.
В крайних случаях рекомендуют ношение ортеза или проводят оперативное вмешательство.
Болезнь Штрюмпеля не представляет никакой опасности для жизни, но из-за нее снижается качество жизни и трудоспособность человека. Однако наследственная выявленная спастическая параплегия имеет свойства к прогрессированию, а признаки могут проявляться в различной степени.
Прогнозировать развитие патологии необходимо только в индивидуальном порядке.
Самое тяжелое течение имеет параплегия Штрюмпеля, начавшаяся в детстве. У пациентов с неосложненной формой, во время пубертатного периода состояние стабилизируется.
Практически все больные могут передвигаться самостоятельно, но некоторые все же теряют эту способность полностью.
Из-за отсутствия четких причин, по которым возник такой синдром, отсутствует и профилактика. Семейная выявленная спастическая параплегия Штрюмпеля – наследственное заболевание.
Все ли корректно в статье с медицинской точки зрения?
Ответьте только в том случае, если у вас есть подтвержденные медицинские знания
Семейный спастический паралич Штрюмпеля
А Б В Г Д Е Ж З И К Л М Н О П Р С Т У Ф Х Ц Ч Ш Щ Э Ю Я
Семейный спастический паралич Штрюмпеля — хроническое прогрессирующее наследственно-дегенеративное заболевание нервной системы, характеризующееся двусторонним поражением пирамидных путей в боковых и передних канатиках спинного мозга. А. Штрюмпель в 1866 г. отметил семейный характер болезни. Применяется также название «семейная спастическая параплегия Эрба–Шарко– Штрюмпеля».
А. Штрюмпель в 1866 г. отметил семейный характер болезни. Применяется также название «семейная спастическая параплегия Эрба–Шарко– Штрюмпеля».
Заболевание является наследственным, чаще передается по аутосомно-доминантному, реже – по аутосомно-рецессивному и сцепленному с полом (с Х-хромосомой) типу.
Патогенез дегенерации и первичный биохимический дефект неизвестны.
Патоморфология. Наиболее часто поражаются поясничная и грудная части спинного мозга, реже – ствол головного мозга. Отмечается симметричное глиозное перерождение пирамидных путей в боковых и передних канатиках, пучках Голля. Описаны случаи дегенеративных изменений в клетках коры передней центральной извилины, передних рогов спинного мозга, мозжечковых проводниках.
Развитие заболевания постепенное. Наиболее часто первые симптомы появляются во втором десятилетии жизни, хотя отмечаются большие колебания возраста, в котором начинается болезнь. Вначале возникают скованность в ногах и быстрая утомляемость при ходьбе, нарастающие по мере прогрессирования заболевания.
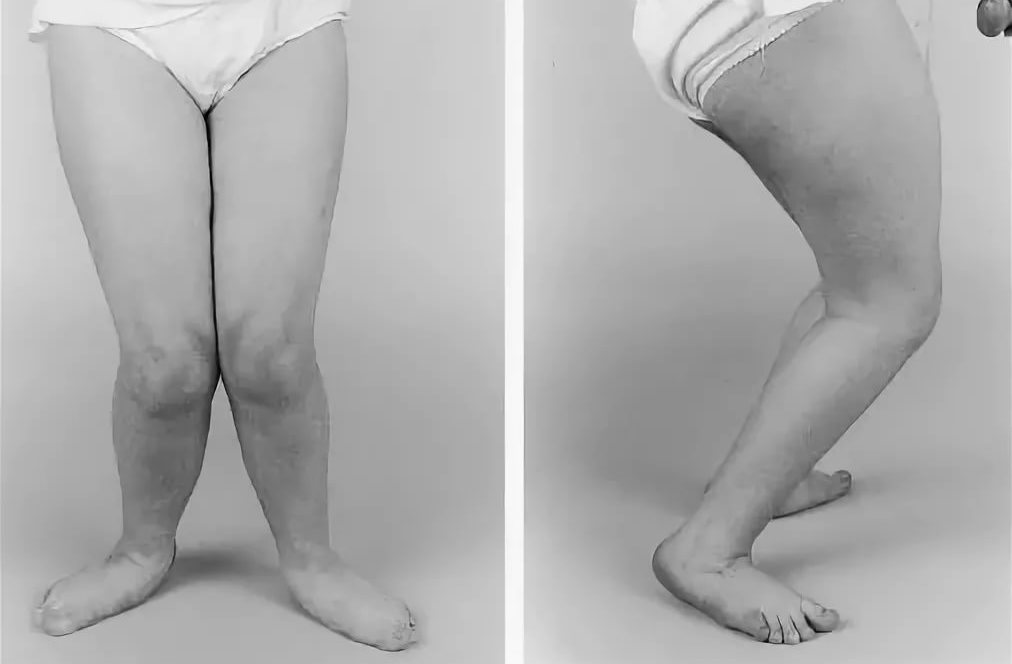
Развивается характерная спастическая по ходка, присоединяются вар усная и эквиноварусная деформации стоп, изменения стоп по типу «стопы Фридрейха», сухожильные и мышечные контрактуры, особенно в голеностопных суставах. Постепенно слабо сть в нижних конечностях нарастает, однако полного паралича нижних конечностей не наблюдается.
При клиническом обследовании больных уже в начальных стадиях заболевания обнаруживается повышение сухожильных рефлексов, рано появляются патологические рефлексы сгибательной и разгибательной групп (Бабинского, Оппенгейма, Россолимо, Гордона, Шеффера, Бехтерева–Менделя, Жуковского), клонусы стоп, коленных чашечек.
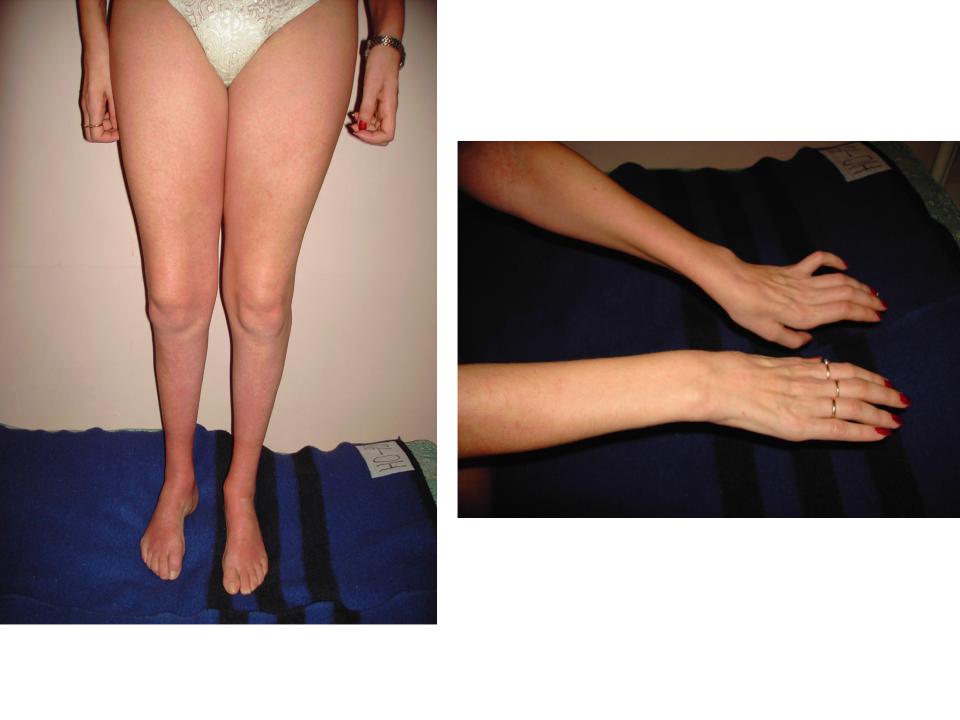
Кожные рефлексы в большинстве случаев сохраняются, функции тазовых органов не нарушены. Расстройства чувствительности отсутствуют. Интеллект сохранен. Значительно позже в патологический процесс вовлекаются верхние конечности.
Нередко к нижнему спастическому парапарезу присоединяются симптомы поражения зрительных и глазодвигательных нервов, нистагм, дизартрия, атаксия и интенционное дрожание.
Диагноз обычно не вызывает затруднений при наличии семейного характера заболевания и типичной клинической картины.
В атипичных спорадических случаях заболевание следует отграничивать от спинальной формы рассеянного склероза, бокового амиотрофического склероза, опухолей спинного мозга и других патологических процессов различной этиологии, вызывающих компрессию спинного мозга, а также фуникулярного миелоза, нейросифилиса и других форм мозжечково-пирамидных дегенерации. Для спинальной формы рассеянного склероза наряду с нижним спастическим парапарезом характерны ремитирующее течение, непостоянство и временная обратимость отдельных симптомов, нарушение функций тазовых органов, выпадение или асимметрия брюшных рефлексов и асимметрия симптомов поражения в целом, изменение иммунологических показателей крови и цереброспинальной жидкости. Решающее значение имеют данные о наследственном характере заболевания. В отличие от бокового амиотрофического склероза болезнь Штрюмпеля начинается в молодом возрасте, отсутствуют признаки поражения периферического мотонейрона (фасцикулярные подергивания, атрофия мелких мышц кисти, характерные изменения ЭМГ), бульбарных расстройств.
 При дифференциации от экстрамедуллярных опухолей и синдрома компрессии спинного мозга другой этиологии имеют значение сегментарные расстройства чувствительности, асимметрия поражения конечностей, наличие блока субарахноидального пространства и белково-клеточная диссоциация в цереброспинальной жидкости при люмбальной пункции, характерные для опухолей. При нейросифилисе в отличие от болезни Штрюмпеля в анамнезе имеются указания на кожные проявления. Ведущими в клинической картине являются симптомы поражения задних канатиков спинного мозга, определяются характерные зрачковые расстройства, изменения в крови, цереброспинальной жидкости.
При дифференциации от экстрамедуллярных опухолей и синдрома компрессии спинного мозга другой этиологии имеют значение сегментарные расстройства чувствительности, асимметрия поражения конечностей, наличие блока субарахноидального пространства и белково-клеточная диссоциация в цереброспинальной жидкости при люмбальной пункции, характерные для опухолей. При нейросифилисе в отличие от болезни Штрюмпеля в анамнезе имеются указания на кожные проявления. Ведущими в клинической картине являются симптомы поражения задних канатиков спинного мозга, определяются характерные зрачковые расстройства, изменения в крови, цереброспинальной жидкости.Дифференциальная диагностика семейной спастической параплегии с другими дегенеративными поражениями спинного мозга бывает иногда затруднительной. Помогает выявление симптомов поражения других отделов нервной системы (мозжечковых, глазных и др.).
Течение заболевания медленно прогрессирующее; отмечается более злокачественное течение при возникновении его в раннем возрасте. При позднем развитии болезни гипертония и гиперрефлексия преобладают над двигательными нарушениями.
При позднем развитии болезни гипертония и гиперрефлексия преобладают над двигательными нарушениями.
Прогноз для жизни благоприятный. Степень утраты трудоспособности зависит от выраженности нарушения функций нервной системы.
Лечение симптоматическое. Назначают препараты, снижающие мышечный тонус, – мидокалм, баклофен, изопротан (скутамил), транквилизаторы: сибазон (седуксен), нозепам (тазепам), хлозепид (элениум). Показаны физиотерапевтические процедуры, парафиновые аппликации на мышцы нижних конечностей.
Применяются точечный массаж, рефлексотерапия, лечебная физкультура, при необходимости – ортопедические мероприятия.
Показаны курсы общеукрепляющего лечения: витамины группы В, метаболические препараты: пирацетам (ноотропил), пиридитол (энцефабол), аминалон, церебролизин, аминокислоты, АТФ, кокарбоксилаза, препараты, улучшающие микроциркуляцию.
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Семейного спастического паралича Штрюмпеля, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику: Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно.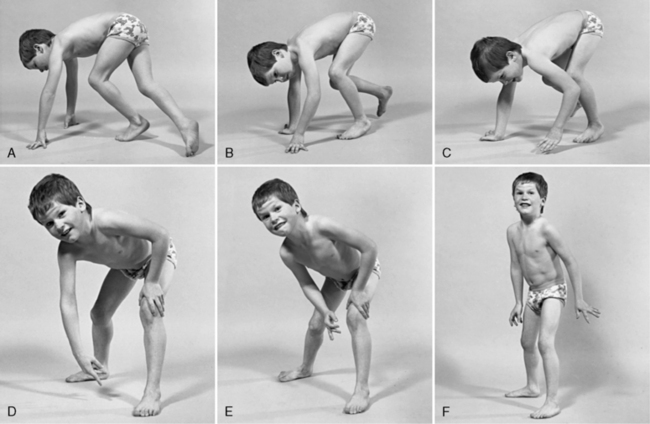
Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом.
Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой.
Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина.
Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь.
Лечение синдрома Штрюмлеля в Израиле
Профессиональное лечение синдрома Штрюмпеля в университетской клинике «Хадасса» осуществляется по самым современным схемам с применением самых актуальных технологических разработок мировой медицины.
Чтобы узнать больше о возможностях клиники «Хадасса» в области лечения синдрома Штрюмпеля, позвоните по указанным телефонным номерам или заполните электронную заявку на лечение.
Почему важно лечить синдром Штрюмпеля?
Синдром Штрюмпеля – тяжелое наследственное заболевание, при котором поражается нервная система человека. Первые проявления болезни возникают ближе к 40 годам, у мужчин куда чаще, чем у женщин. При синдроме больной жалуется на скованность, усталость в ногах. Постепенно симптомы нарастают, нарушается нормальная функция органов малого таза. Доктора могут отметить изменившуюся походку, слабость мышц, деформацию стоп.
Доктора могут отметить изменившуюся походку, слабость мышц, деформацию стоп.
Как диагностируют синдром Штрюмпеля в клинике «Хадасса»?
Несмотря на некоторые красноречивые сигналы, такие как жалоба на невозможность оторвать стопы от пола, синдром Штрюмпеля подтверждается только одним достоверным способом – ДНК- диагностикой. В клинике «Хадасса» пациент может пройти все необходимые обследования для того, чтобы выявить скрытую болезнь, или узнать о прогрессе проявившейся, всего за несколько дней.
Первый этап. Визит в клинику
Для того чтобы поставить диагноз «синдром Штрюмпеля», пациенту нужно пройти консультацию у опытного невролога. Доктор проведет осмотр, проведет специфические тесты. Например, тест на рефлекс Штрюмпеля, который заключается в специфическом изгибе большого пальца ноги при попытке пациента согнуть ногу в колене, когда врач оказывает незначительное противодействие. Также доктор выдает направления на дополнительные исследования.
Второй этап. Диагностика
Для того чтобы подобрать оптимальное лечение, врачи должны убедиться, что пациент не страдает от схожих болезней – рассеянного склероза, опухолей спинного мозга, миелопатии и так далее. Для этого проводятся следующие тесты:
Магнитно-резонансная томография. Это современное исследование поможет выявить умеренную неспецифическую атрофию спинного мозга;
Лабораторные анализы крови, мочи, спиномозговой жидкости;
ДНК-тест.
Третий этап. Подбор методики лечения
После подтверждения диагноза доктора анализируют данные и состояние больного и подбирают оптимальный метод лечения симптомов синдрома Штрюмпеля.
Как лечат синдром Штрюмпеля в клинике «Хадасса»?
Так как синдром является наследственным генетическим заболеванием, лечение может быть только симптоматическим. При это прогноз у болезни благоприятный, если симптоматическая терапия подобрана профессионально. В клинике «Хадасса» пациентам с синдромом Штрюмпеля предлагают следующие методы лечения:
В клинике «Хадасса» пациентам с синдромом Штрюмпеля предлагают следующие методы лечения:
Медикаментозная терапия. Для улучшения состояния больного назначаются различные препараты. Многие израильские лекарства не имеют аналогов и оказывают значительный лечебный эффект;
Инъекции ботулинического токсина. Подобные действия сродни тому, как для предотвращения появления морщин женщины делают уколы ботоксом. В случае с синдромом Штрюмпеля уколы нужны для предотвращения развития болезни;
Физиотерапия. Реабилитологи клиники «Хадасса» подбирают для пациента оптимальные физические упражнения и физиотерапевтические процедуры, которые помогут сохранить подвижность и крепость мышц.
Позвоните или пишите в клинику «Хадасса» в любое удобное для вас время, чтобы наши консультанты смогли рассказать вам все необходимое о порядке лечения, ценах и условиях медицинского туризма в Израиле.
причины, симптомы, диагностика, лечение, профилактика
Представляет с собой форму семейной спастической параплегии, характеризующуюся развитием наследственной дегенеративной миелопатии, обусловленной двусторонним поражением боковых и передних спинномозговых столбов, чаще всего, развивающемся на поясничном уровне.
Случай семейной спастической параплегии был впервые описан в 1883 г. немецким клиницистом А. Штрюмпелем, позже изучением патологии занимался М. Лорен. Именно в честь этих исследователей заболевание было названо болезнь Штрюмпеля-Лорена. В современной медицине чаще всего заболевание упоминается как болезнь Штрюмпеля. Патогенетическим фактором недуга являются прогрессирующее глиальное перерождение пирамидных трактов передних и боковых столбов на уровне грудных и поясничных сегментов спинного мозга. Параллельно с этим у больного может отмечаться развитие атрофических процессов в передних рогах, дегенерационное поражение проводящих путей мозжечка, уменьшение количества нейронов моторной зоны коры, глиоз пирамидных трактов на уровне ствола мозга.
Возрастной период манифестации клинической картины заболевания достаточно широк: от 1 до 80 лет. При этом чаще всего дебют недуга припадает на возраст от 10 до 30 лет. Благодаря возможностям современной генетики в последнее время удалось выявить множество генетических вариантов семейной спастической параплегии. В настоящий момент выделяют примерно 17 хромосомных локусов, мутации в которых приводят к развитию болезни Штрюмпеля с аутосомно-доминантным наследованием, 29 локусов, которые отвечают за аутосомно-рецессивный тип наследования и 4 локуса, которые обусловлены с наследованием сцепленным с Х хромосомой.
В настоящий момент выделяют примерно 17 хромосомных локусов, мутации в которых приводят к развитию болезни Штрюмпеля с аутосомно-доминантным наследованием, 29 локусов, которые отвечают за аутосомно-рецессивный тип наследования и 4 локуса, которые обусловлены с наследованием сцепленным с Х хромосомой.
Ранним проявлением заболевания является задержка формирования навыков ходьбы, а также хождения на цыпочках. У более взрослых больных заболевание манифестирует появлением затруднений при ходьбе, что обусловлено частыми падениями. В начале заболевания пациенты могут жаловаться на плохую опору на стопу при ходьбе и скованность в ногах. У таких детей определяется в ногах повышенный мышечный тонус. При манифестации недуга гипертонус может носить переходящий характер и усиливаться при ходьбе и исчезать во время отдыха. Спастичность более выражена в камбаловидных мышцах голени, приводящих и задних мышцах бедра. Иногда спастичность может быть ассиметричной. Некоторые больные жалуются на проблемы лишь с одной ногой, но при неврологическом осмотре выявляется двустороннее повышение тонуса и гиперрефлексия в обеих ног с наличием пирамидных стопных знаков.
Некоторые больные жалуются на проблемы лишь с одной ногой, но при неврологическом осмотре выявляется двустороннее повышение тонуса и гиперрефлексия в обеих ног с наличием пирамидных стопных знаков.
Для болезни Штрюмпеля характерно медленно прогрессирующее течение. Снижение силы в мышцах ног появляется спустя длительное время с момента поражения. У некоторых больных отмечается развитие сенсорных расстройств, проявляющихся легкими нарушениями вибрационного восприятия, иногда у больных возникают парестезии в голенях и стопах. Более тяжелые нарушения чувствительности возникают при присоединении полиневропатии или осложненном течении заболевания.
Как правило, атрофические изменения мышц ног, появляются на поздних стадиях болезни Штрюмпеля и характеризуются развитием обездвиженности обусловленной тяжелыми парезами. Некоторые формы семейной наследственной параплегии могут сопровождаться атрофией мышц рук. На последних стадиях заболевания может возникнуть спастический парез верхних конечностей и недержание мочи. Тяжелые формы заболевания сопровождаются развитием неврологической симптоматики, когнитивными нарушениями: от легкой олигофрении до тяжелой деменции. У некоторых больных отмечается развитие эпилепсии, оптической нейропатии, врожденной ретинопатии, дизартрии и мозжечкового синдрома.
Тяжелые формы заболевания сопровождаются развитием неврологической симптоматики, когнитивными нарушениями: от легкой олигофрении до тяжелой деменции. У некоторых больных отмечается развитие эпилепсии, оптической нейропатии, врожденной ретинопатии, дизартрии и мозжечкового синдрома.
Особое диагностическое значение имеет наличие у больного нижней центральной параплегии и семейный характер заболевания. Для подтверждения диагноза больному назначается магниторезонансная или компьютерная томография.
Вспомогательным методом в диагностике болезни Штрюмпеля является электронейромиография и исследование вызванных потенциалов. При помощи данного обследования удается определить наличие и степень нейропатии.
ЛечениеВ основе лечения лежит использование миорелаксантов и транквилизаторов, которые также оказывают расслабляющее на мышцы воздействие. Лечение начинают с минимальных доз, при постепенном увеличении до достижения необходимого терапевтического эффекта.
Если пероральный прием не позволяет добиться желаемого эффекта, препараты вводят внутримышечно. Возможно эндолюмбальное локальное введение. При грубой спастике прибегают к установке помпы для постоянной интратекальной инфузии баклофена. Указанное лечение является симптоматическим и не позволяет полностью излечиться.
Иногда для уменьшения спастики практикуют введение ботулотоксина в задние мышцы бедер и икроножные мышцы.
ПрофилактикаПока не разработано эффективных методов профилактики развития болезни Штрюмпеля.
Анкилозирующий спондилит (болезнь Мари-Штрумпеля, болезнь Бехтерева)
.
Анкилозирующий спондилит (АС) — хроническое воспалительное заболевание позвоночника и крестцово-подвздошных суставов. Это может быть связано с рядом внесуставных структур, таких как глаз, желудочно-кишечный тракт (ЖКТ), кожа, легкие, почки и сердце. АС обычно начинается во втором или третьем десятилетии, когда соотношение мужчин и женщин составляет от 2: 1 до 3: 1.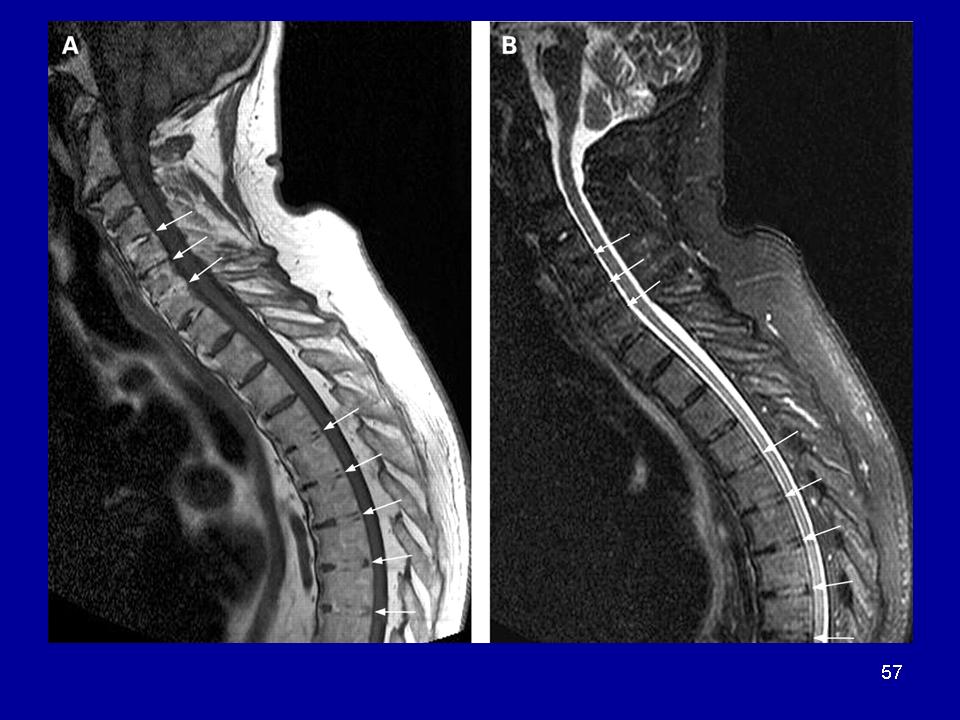 АС является прототипом спондилоартрита.Двумя основными типами спондилоартрита являются AS (осевой спондилоартрит с сакроилитом на простом рентгеновском снимке) и нерадиографический спондилоартрит (без сакролилита на простом рентгеновском снимке). Термин аксиальный спондилоартрит все чаще используется для обозначения АС.
АС является прототипом спондилоартрита.Двумя основными типами спондилоартрита являются AS (осевой спондилоартрит с сакроилитом на простом рентгеновском снимке) и нерадиографический спондилоартрит (без сакролилита на простом рентгеновском снимке). Термин аксиальный спондилоартрит все чаще используется для обозначения АС.
У пациентов с АС наблюдается скованность по утрам и боли в пояснице или боли в ягодицах. Этиология остается неясной, но, по-видимому, является иммуноопосредованной и связана с сильным наследственным компонентом: лейкоцитарным антигеном человека (HLA) B27.
Энтезит, воспаление прикрепленных сухожилий и связок, является одним из основных признаков АС и приводит ко многим из симптомов боли.
AS диагностируется на основании клинических, лабораторных и визуализационных данных, характерных для AS. Вероятность диагноза варьируется в зависимости от имеющихся конкретных результатов. Не существует конкретного теста, единственного исторического признака или результатов визуализации, которые были бы достаточны для диагностики или исключения болезни.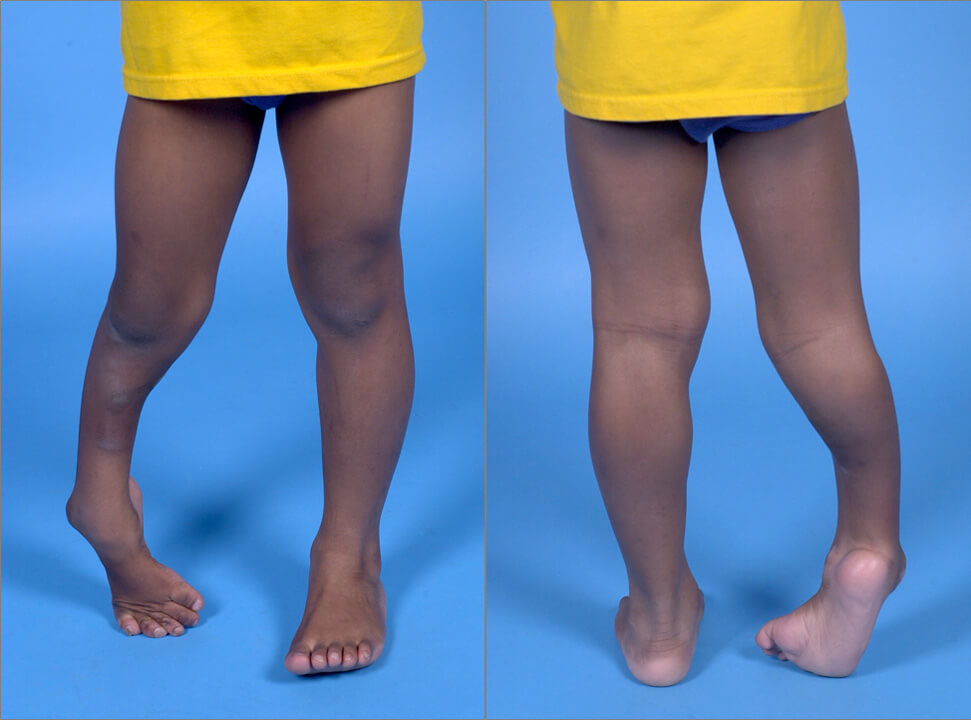
Модифицированные Нью-Йоркские критерии для AS, предложенные в 1984 году, следующие:
Боль в пояснице продолжительностью не менее 3 месяцев, которая уменьшается после упражнений и не уменьшается после отдыха.
Ограничение движений поясничного отдела позвоночника в сагиттальной и фронтальной плоскостях.
Увеличение груди, уменьшенное относительно нормальных значений для возраста и пола.
Рентгенологический односторонний сакроилеит 3-4 степени.
Рентгенологический двусторонний сакроилеит 2-4 степени.
Диагноз СА является определенным, если присутствует 4 или 5 и любой из критериев 1–3.
Нью-Йоркские критерии наиболее полезны для диагностированного заболевания на более поздних стадиях, поскольку они в значительной степени зависят от рентгенологической диагностики сакроилеита. Полезность этих критериев снижается у пациентов с ранним заболеванием, которые могут не иметь четких рентгенологических данных.
Важно установить диагноз раннего АС до развития необратимой деформации.В 2009 году Международное общество по оценке спондилоартрита (ASAS) предложило новые критерии аксиального спондилоартрита. Эти критерии применимы к людям с болью в спине в течение 3 месяцев и более с возрастом начала <45 лет или меньше.
КритерииASAS также включают признаки спондилоартрита, HLA B-27 и CRP / СОЭ.
Особенности SpA (спондилоартрита)
Воспалительная боль в спине
3 месяца и более
Коварное начало
Без разгрузки с остатком
Улучшение с упражнением
Боль ночью
Боль в пятке вследствие тендинита ахиллова сухожилия или подошвенного фасциита (энтезита)
Дактилит (колбасный палец; болезненное воспаление пальца)
Увеит
Семейная история AS
Альтернативная боль в ягодицах
Псориаз
Асимметричный артрит
Положительный ответ на НПВП
Высокая СОЭ или CRP
ВЗК (болезнь Крона или язвенный колит)
Сакроилит при визуализации PLUS Один или несколько признаков SpA
Активное воспаление подвздошного сустава на МРТ, указывающее на SpA, или HLA B -27 Plus 2, или более признаков SpA и / или определенный сакроилит, согласно модифицированным Нью-Йоркским критериям, особенности позвоночника с AS почти никогда не проявляются до достижения возраста 16.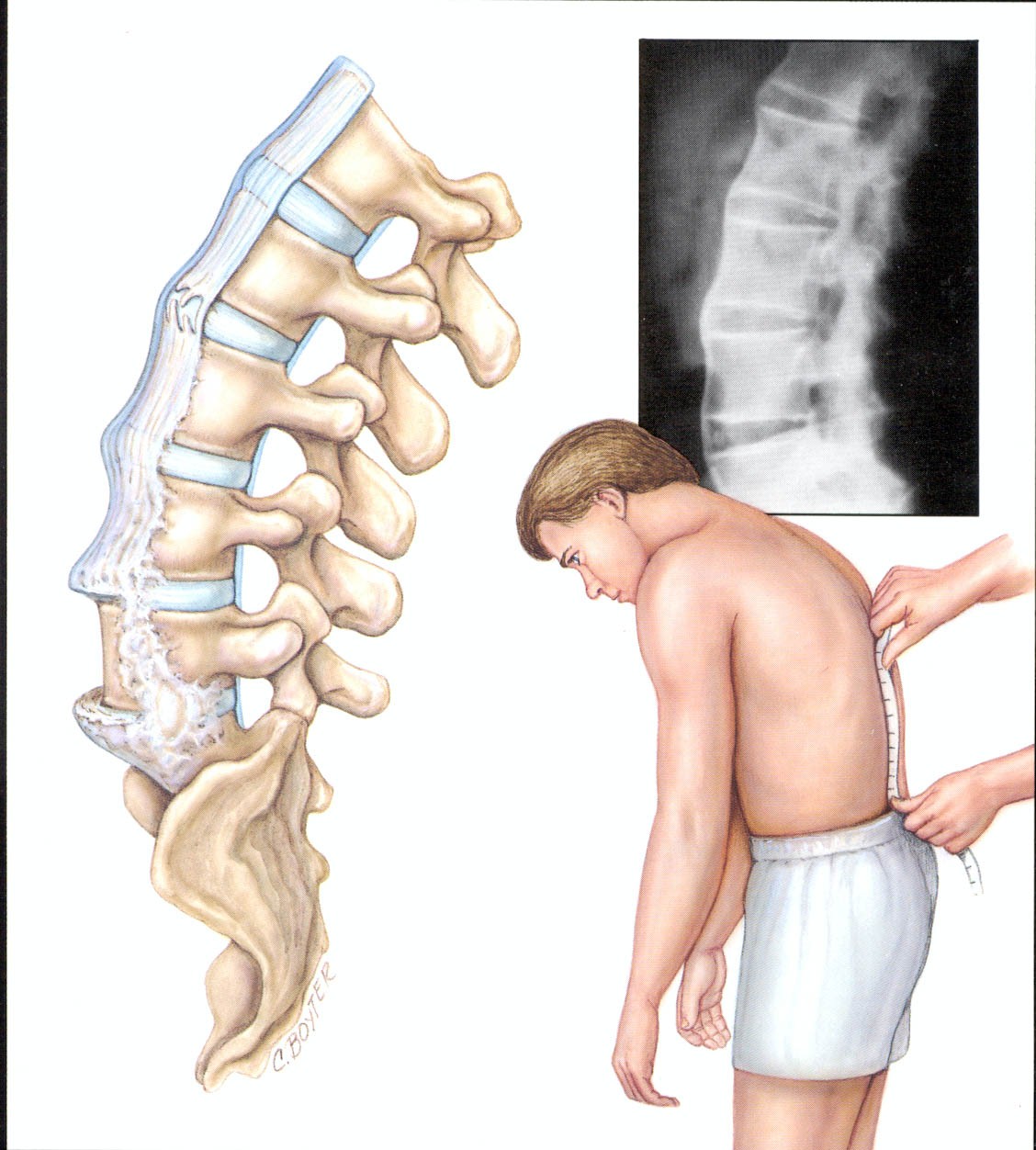 Менее 5% пациентов имеют симптомы, которые начинаются после 40 лет, хотя АС часто диагностируется после 40 лет, поскольку начальные симптомы незначительны или игнорируются. Средний возраст начала составляет 26 лет, и большинство симптомов, как правило, проявляются в течение третьего десятилетия.
Менее 5% пациентов имеют симптомы, которые начинаются после 40 лет, хотя АС часто диагностируется после 40 лет, поскольку начальные симптомы незначительны или игнорируются. Средний возраст начала составляет 26 лет, и большинство симптомов, как правило, проявляются в течение третьего десятилетия.
Типичный пациент с АС жалуется на тупую боль в нижней части поясницы или ягодичной области. Они могут жаловаться на утреннюю скованность в пояснице, которая сохраняется в течение нескольких часов и улучшается при физической активности. Начало обычно коварное.Боль, как правило, сохраняется более 3 месяцев и заметно усиливается после отдыха и уменьшается после упражнений. Ночные обострения боли часто заставляют пациента вставать и двигаться.
Болезненность костей, вероятно, из-за энтезита, часто встречается на таких участках, как реберно-грудные сочленения, остистые отростки, гребни подвздошных костей, большие вертела, седалищные бугры, бугорки большеберцовой кости и пятки. Дополнительные данные включают синовит тазобедренных, коленных, голеностопных и плюснево-фаланговых (MTP) суставов.Суставы верхних конечностей, кроме плечевого, практически не затрагиваются.
Дополнительные данные включают синовит тазобедренных, коленных, голеностопных и плюснево-фаланговых (MTP) суставов.Суставы верхних конечностей, кроме плечевого, практически не затрагиваются.
Артрит бедер и плеч встречается у 30% пациентов с АС. Артрит периферических суставов, кроме бедер и плеч, обычно асимметричный, встречается у 30% пациентов. Боль и скованность в шее обычно являются поздними проявлениями. Периферический артрит преходящий в 50% случаев и постоянный в 25% случаев. Передний увеит развивается примерно у 33% пациентов с АС и может возникать до начала спондилита.После того, как диагностирован АС, увеит обычно не совпадает с обострением артрита.
Существует четкая взаимосвязь между воспалением кишечника и сакроилеитом. Хотя до 25% пациентов с болезнью Крона или язвенным колитом (ЯК) страдают сакроилеитом, почти у 60% пациентов с АС наблюдаются субклинические признаки воспалительных изменений в тонкой или толстой кишке. Около 10% пациентов с АС имеют явную болезнь Крона или ЯК.
У нескольких процентов пациентов с АС развиваются нарушения сердечной проводимости или аортит.Риск этих изменений увеличивается с возрастом и продолжительностью заболевания, но может проявляться полной блокадой сердца и аортальной недостаточностью с застойной сердечной недостаточностью.
Легочные признаки у пациентов с АС включают фиброз верхней доли и внелегочное рестриктивное заболевание легких из-за повышенной жесткости грудной стенки. Фиброз наблюдается только при длительном заболевании и обнаруживается только у 1% пациентов с АС. Внелегочная рестриктивная болезнь легких обычно компенсируется увеличением экскурсии диафрагмы и редко приводит к тяжелым легочным симптомам, но может усугубить другие формы легочной болезни.
Неврологическое заболевание обычно возникает из-за поражения спинного мозга или нервных корешков в результате перелома позвоночника. До 25% пациентов с АС могут иметь псориатические поражения.
Распространенность АС сильно варьируется. По оценкам, он составляет примерно 1% среди кавказского населения в Соединенных Штатах и Соединенном Королевстве, но данных немного. Среди коренных американцев хайда в Северной Канаде распространенность составляет около 6%.
По оценкам, он составляет примерно 1% среди кавказского населения в Соединенных Штатах и Соединенном Королевстве, но данных немного. Среди коренных американцев хайда в Северной Канаде распространенность составляет около 6%.
AS хорошо коррелирует с антигеном гистосовместимости HLA-B27.Более 90% пациентов с АС унаследовали HLA-B27. Среди кавказцев Северной Америки распространенность HLA-B27 составляет 7%. По оценкам, из тех, кто унаследовал HLA-B27, только у 1-6% действительно развивается AS. Поэтому HLA B-27 не используется в качестве диагностического теста. В семьях пациентов с АС распространенность может составлять почти 30% у родственников первой степени родства с HLA-B27. Уровень конкордантности среди однояйцевых близнецов составляет 65%, что подтверждает предполагаемый генетический компонент.
AS чаще встречается у мужчин, чем у женщин.Соотношение мужчин и женщин оценивается в 2: 1, хотя считается, что женщины с СА недооцениваются.
.
Воспалительное заболевание кишечника
Псориаз
Реактивный артрит
Ревматоидный артрит
Остеоартроз
Тендинит
Подошвенный фасциит
Фибромиалгия
Компрессионный перелом позвонка
Инфекция крестцово-подвздошного сустава
Семейная средиземноморская лихорадка
AS отличается появлением симптомов в молодом возрасте.Это может быть связано с симптомами и особенностями воспалительного заболевания кишечника и псориаза. Средний возраст дебюта — 26 лет. Ключевым историческим компонентом СА является то, что боль в спине сохраняется более 3 месяцев, усиливается после отдыха и уменьшается после упражнений.
.
Как признак сакроилеита, некоторые пациенты могут испытывать боль в ягодицах при надавливании на крестец. У многих пациентов ограничена подвижность позвоночника. Это можно увидеть при сгибании и разгибании поясничного отдела позвоночника.Модифицированный тест Шобера может использоваться для определения сгибания поясничного отдела позвоночника.
У многих пациентов ограничена подвижность позвоночника. Это можно увидеть при сгибании и разгибании поясничного отдела позвоночника.Модифицированный тест Шобера может использоваться для определения сгибания поясничного отдела позвоночника.
Модифицированный тест Шобера выполняется путем нанесения двух горизонтальных линий на 5 см ниже и 10 см выше пояснично-крестцового перехода. Пояснично-крестцовый переход, определяемый горизонтальной линией между задне-верхними подвздошными ости. Стоя прямо, пятки плотно прижаты друг к другу, пациент наклоняется как можно дальше вперед с полностью вытянутыми коленями и измеряет расстояние между двумя линиями. Если расстояние увеличивается на 5 см, это нормально.Если расстояние увеличивается менее чем на 4 см, это ненормально и является признаком ограниченной подвижности позвоночника.
Признаки синовита и энтезита также являются обычными. Это часто проявляется болезненностью суставов или костной болезнью в местах прикрепления сухожилий.
В остальном медицинский осмотр на AS сильно варьируется.
Ни один лабораторный тест не является диагностическим для AS. СРБ и СОЭ часто, но не всегда повышены. В настоящее время это остается клиническим диагнозом, хотя визуализация для установления сакроилеита полезна.
Ниже приведены этапы диагностики, адаптированные из модифицированного Берлинского алгоритма ASAS 2013 года, чтобы помочь установить диагноз.
Этап 1. У пациента с хронической болью в спине (не менее 3 месяцев) и возрастом <45 лет сделать рентгеновский снимок таза для исследования подвздошных суставов. Диагноз АС может быть установлен, если он соответствует модифицированным критериям Нью-Йорка. Дальнейшего расследования не требуется.
Этап 2 — У пациента с отрицательным результатом рентгеновского снимка SI-сустава просмотрите историю болезни на предмет 11 признаков SpA.Наличие 4 из 11 характерных признаков SpA является диагностическим признаком нерадиографического аксиального спондилоартрита (nr-AxSpA). Никакого дополнительного тестирования не требуется.
Этап 3 — У пациента с менее чем 4 признаками SpA и отрицательным рентгеновским снимком SI-сустава проводится тестирование HLA B-27. Положительный тест на HLA-B27 обычно может указывать на диагноз нерадиографического аксиального спондилоартрита (nr-AxSpA).
Однако у пациента с 2 или 3 признаками, отрицательным HLA-B27 и отсутствием сакроилеита на рентгеновском снимке вероятность СПА низкая.Если клиническое подозрение остается высоким, для подтверждения диагноза nr-AxSpA необходимо провести МРТ SI-суставов.
Шаг 4 — Пациенту с нулевым или одним признаком SpA, но + HLA-B27 необходимо сделать МРТ суставов SI. МРТ с положительным результатом на сакроилеит подтверждает диагноз нерадиографического спондилоартрита.
Диагноз AS — это клинический диагноз. Роль HLA-B27 в установлении диагноза неясна и исследуется.HLAB27 полезен для пациентов со средней вероятностью. Скорость оседания эритроцитов (СОЭ) и С-реактивный белок (СРБ) могут использоваться для определения активности заболевания, но не являются диагностическими. Щелочная фосфатаза в сыворотке часто повышается при запущенном заболевании. Повышенный уровень IgA является обычным явлением. Ревматоидный фактор, антитела против CCP, ANA и ANCA обычно находятся в пределах нормы и могут быть ненормальными, если эти заболевания сосуществуют.
На переднезадних рентгенограммах таза может быть обнаружено псевдорасширение крестцово-подвздошного сустава, а также склероз, эрозии и анкилозирование.У пациента с высоким клиническим подозрением и отрицательным результатом рентгенологического исследования тазовых органов наиболее чувствительны МРТ с Т1-взвешенными последовательностями плюс STIR или Т2-взвешенные изображения с подавлением жира. Отек костного мозга в субхондральных областях на STIR или на T2-взвешенных изображениях с подавлением жира является определяющим наблюдением активного воспаления SI-суставов. Эти данные МРТ сами по себе не являются диагностическими и должны интерпретироваться в контексте других клинических и лабораторных данных. Повышение контрастности не требуется.
КТ-сканированиеимеет очень ограниченное применение, поскольку оно неспецифично и не дает информации о воспалении суставов. Это также приводит к большему облучению.
.
КТ, сканирование костей
.
Активность заболевания следует определять с помощью индекса активности заболевания анкилозирующим спондилитом (BASDAI). В отличие от BASDAI, оценка активности болезни при анкилозирующем спондилите (ASDAS) учитывает реактанты острой фазы.Общая оценка должна включать физическую функцию, боль, подвижность позвоночника, жесткость, утомляемость, заболевание периферических суставов, общую оценку состояния пациента и реактивы острой фазы. Первоначальное лечение всех пациентов должно включать физиотерапию, упражнения и обучение пациентов.
Всем пациентам с симптомами следует назначить пробную терапию нестероидными противовоспалительными препаратами (НПВП) для облегчения скелетно-мышечных симптомов. Чтобы оценить эффективность НПВП, его следует назначать в постоянной дозе на регулярной основе в течение не менее 4 недель.Рекомендуется попробовать как минимум два разных НПВП, прежде чем объявить этот класс препаратов неэффективным.
Другие анальгетики и опиоиды редко оказываются эффективными во время активного АС, когда используются отдельно.
Пациенты с показателем BASDAI 4 или выше и общим баллом врача 2 или выше и заболеванием, устойчивым к НПВП, считаются умеренными и должны получать дополнительную терапию.
Сульфасалазин (от 2 до 3 г в день) может принести пользу пациентам, главным образом с периферическим артритом.Пациентам с аксиальными симптомами следует назначать средство против фактора некроза опухоли (анти-TNF), такое как этанерцепт, или моноклональные антитела, такие как инфликсимаб, голимумаб или адалимуубаб. Для пациентов с АС и воспалительным заболеванием кишечника моноклональные антитела предпочтительнее этанерцепта.
Модифицирующие заболевание противоревматические препараты (DMARD), за исключением сульфасалазина, упомянутого выше, не рекомендуются для лечения анкилозирующего спондилита в соответствии с рекомендациями Международного общества по оценке спондилоартрита (ASAS).
Кортикостероиды оказывают минимальное влияние на артрит, особенно спондилит, и могут усугубить остеопению.
Метотрексат и лефлуномид не помогают при этом заболевании.
Моноклональное антителотоцилизумаба против рецептора ИЛ-6 и абатацепта, которое блокирует костимуляцию Т-клеток, не оказалось полезным.
Секукинумаб, моноклональное антитело к интерлейкину (IL) -17A, играет полезную роль в активном AS у пациентов, у которых наблюдается неадекватный ответ на другие методы лечения, включая ингибиторы TNF.
Устекинумаб представляет собой моноклональное антитело против белковой субъединицы p40, общей для IL-12, IL-23.
Было показано, чтоАмитриптилин приносит пользу пациентам с АС, нарушением сна и утомляемостью.
Памидронат (60 мг ежемесячно внутривенно) снижает активность заболевания и вызывает ощутимое улучшение функции.
.
Острый передний увеит является наиболее частым внесуставным проявлением АС и может быть основным симптомом заболевания.Увеит можно лечить с помощью местных глюкокортикоидов и мидриатических средств. Иногда в рефрактерных случаях могут потребоваться системные кортикостероиды или иммунодепрессанты.
.
Активность заболевания следует определять с помощью BASDAI. Физическую функцию можно определить с помощью функционального индекса при анкилозирующем спондилите (BASFI).
.
СОЭ и СРБ можно использовать для отслеживания активности заболевания.
.
Долгосрочное лечение всех пациентов должно включать физиотерапию, упражнения и обучение пациентов.Продолжительные периоды иммобилизации должны быть сведены к минимуму.
Настоятельно рекомендуется бросить курить.
Иммунодепрессанты, такие как этанерцепт и инфликсимаб, были связаны с инфекциями (туберкулезом), панцитопенией, демиелинизирующим заболеванием, обострениями застойной сердечной недостаточности и реакциями в месте инъекции. При приеме этих лекарств также может наблюдаться лекарственная волчанка.
Памидронат может вызвать преходящую лимфопению.
Следует избегать длительного применения НПВП у пациентов с хроническим заболеванием почек, недавней язвенной болезнью в анамнезе и умеренным риском ишемической болезни сердца.
НПВП следует использовать вместе с метотрексатом. НПВП могут повышать уровень метотрексата в сыворотке крови.
.
Избегайте НПВП и метотрексата.
.
С осторожностью при использовании средств против TNF, особенно у пациентов, являющихся хроническими носителями гепатита B.
Агенты против TNF были связаны с обострениями застойной сердечной недостаточности. Рекомендуется соблюдать осторожность.
Избегайте длительного приема НПВП.
Осторожно при использовании системных глюкокортикоидов, которые обычно не рекомендуются для лечения АС.
Агенты против TNF связаны с повышенным риском лимфомы.
.
Осторожно при использовании дополнительных иммунодепрессантов, особенно препаратов против TNF.
Без изменений в стандартном управлении.
Избегайте применения НПВП у пациентов с недавними кровотечениями из верхних отделов ЖКТ.
Агенты против TNF и метотрексат могут вызывать угнетение костного мозга.
Без изменений в стандартном управлении.
.
Наблюдать за реакциями в месте инфузии или побочными эффектами препаратов против TNF.
.
В большинстве случаев это заболевание лечится амбулаторно. Продолжительность стационарного лечения неясна.
.
НЕТ
Рекомендуется тщательное наблюдение ревматолога.
.
.
НЕТ
.
НЕТ
.
Если АС достаточно тяжелый, чтобы потребовать госпитализации, может потребоваться стационарная физиотерапия, если состояние пациента быстро не улучшится.Может потребоваться медицинское обследование и реабилитация. Если пациент сильно ослаблен, может быть целесообразным его размещение в доме престарелых. Функциональная оценка и очищенное производное белка (PPD) обычно требуется для помещения в дом престарелых.
.
НЕТ
.
НЕТ
.
НЕТ
Сориано, Э., Клегг, Д., Лисс, Дж. «Критическая оценка рекомендаций по лечению анкилозирующего спондилита: антиревматические препараты, изменяющие заболевание». Am J Med Sci. об. 5. 2012. С. 357-359.
Рен, Л., Ли, Дж., Ренкуан, Т. «Эффективность агентов противоопухолевого фактора некроза (α) у пациентов с анкилозирующим спондилитом». Am J Med Sci. об. 6. 2013. С. 455-461.
Рудвалейт, М. «Разработка критериев классификации аксиального спондилоартрита Международным обществом по оценке спондилоартрита (часть II): проверка и окончательный выбор». Ann of Rheum Dis. об. 68. 2009. С. 777
.Битая, Д.«Моноклональные антитела к интерлейкину-17A секукинумаб в лечении анкилозирующего спондилита: рандомизированное двойное слепое плацебо-контролируемое исследование». Ланцет. об. 382. 2013. С. 1705
.Поддубный, Д. «Устекинумаб для лечения больных активным анкилозирующим спондилитом». Ann Rheum Dis. об. 73. 2014. С. 817
.Copyright © 2017, 2013 ООО «Поддержка принятия решений в медицине». Все права защищены.
Ни один спонсор или рекламодатель не участвовал, не одобрял и не платил за контент, предоставляемый Decision Support in Medicine LLC.Лицензионный контент является собственностью DSM и защищен авторским правом.
Наследственная спастическая параплегия — NHS
Наследственная спастическая параплегия — это общий термин для группы редких наследственных заболеваний, вызывающих слабость и жесткость мышц ног. Со временем симптомы постепенно ухудшаются.
Он также известен как семейный спастический парапарез или синдром Штрюмпелла-Лоррена.
Трудно точно определить, у скольких людей есть наследственная спастическая параплегия, потому что ее часто неправильно диагностируют.
По оценкам, от 1 из 11 000 до 1 из 77 000 человек.
Симптомы наследственной спастической параплегии
Степень тяжести и прогрессирование симптомов зависит от человека.
Около 90% людей с наследственной спастической параплегией имеют так называемую «чистую форму» этого заболевания.
Это означает, что их симптомы в основном ограничиваются слабостью нижних конечностей, непроизвольными спазмами и ригидностью мышц (спастичностью).
Остальные 10% имеют сложную или сложную форму заболевания.
Это означает, что у них есть другие симптомы, помимо мышечной слабости и спастичности.
У них может быть широкий спектр симптомов.
Чистая наследственная спастическая параплегия
Основные симптомы чистой наследственной спастической параплегии:
- Постепенная слабость в ногах
- Повышение мышечного тонуса и жесткости (спастичность)
- Проблемы с мочеиспусканием — например, острая необходимость в мочеиспускание, даже если мочевой пузырь не заполнен
- Отсутствие чувствительности в стопах (иногда)
У детей может развиться скованность ног и проблемы при ходьбе, например спотыкание и спотыкание, особенно на неровной поверхности.
Это потому, что им трудно сгибать пальцы ног вверх из-за слабых мышц бедра.
Некоторым людям со временем может потребоваться трость или инвалидное кресло, чтобы помочь им передвигаться.
Другим может не потребоваться какое-либо мобильное оборудование.
Осложненная наследственная спастическая параплегия
При осложненной наследственной спастической параплегии дополнительные симптомы могут включать:
- Повреждение нервов стоп или других конечностей (периферическая невропатия)
- эпилепсия
- Проблемы с равновесием, координацией и речью (атаксия)
- Проблемы со зрением, такие как повреждение сетчатки (ретинопатия) и повреждение зрительного нерва (оптическая нейропатия)
- слабоумие
- ихтиоз — заболевание, вызывающее широко распространенную и стойкую толстую и сухую кожу «рыбьей чешуи».
- Задачи обучения и развития
- потеря слуха
- Проблемы с речью, дыханием или глотанием
Что вызывает наследственную спастическую параплегию?
Большинство людей с чистой наследственной спастической параплегией унаследовали дефектный ген от одного из своих родителей.
Люди с осложненной формой заболевания обычно наследуют дефектный ген от обоих родителей.
Аномалия гена вызывает повреждение длинных нервов в позвоночнике.
Эти нервы обычно контролируют мышечный тонус и движение в нижней части тела.
Диагностика наследственной спастической параплегии
Наследственная спастическая параплегия диагностируется после тщательного клинического обследования и выявления типичных симптомов.
В первую очередь необходимо исключить другие состояния, вызывающие проблемы с подвижностью, ригидность и слабость мышц, такие как рассеянный склероз и церебральный паралич.
При диагностике можно использовать ряд специализированных тестов, включая МРТ головного и спинного мозга, анализ спинномозговой жидкости, тесты нервной проводимости и ЭМГ.
В некоторых случаях также может потребоваться генетическое тестирование.
Лечение наследственной спастической параплегии
Невозможно предотвратить, замедлить или обратить вспять наследственную спастическую параплегию, но с некоторыми симптомами можно справиться, что облегчит повседневную деятельность.
Например:
- миорелаксанты, такие как инъекции баклофена, тизандина и ботулина (ботокса), могут использоваться для облегчения спастичности
- Регулярная физиотерапия важна для улучшения и поддержания мышечной силы и диапазона движений
- трудотерапия может помочь человеку легче выполнять свою повседневную деятельность и восстановить максимальную независимость
- Ортез на голеностопный сустав можно носить на голени, чтобы помочь выпрямить и контролировать голеностопный сустав и стопу, а также улучшить ходьбу
- Иногда может потребоваться операция для освобождения сухожилий или укороченных мышц
Осложнения наследственной спастической параплегии
Возможные осложнения наследственной спастической параплегии включают:
- сокращение и укрепление икроножных мышц — регулярная физиотерапия может помочь предотвратить это
- Холодные ноги — это довольно частое явление, возникающее в результате повреждения нервов в позвоночнике
- крайняя усталость (утомляемость) — это может быть из-за дополнительных усилий, необходимых для ходьбы, и симптомов, мешающих спать
- Боль в спине и коленях — вызванная мышечной слабостью и проблемами при ходьбе
- стресс и депрессия
Outlook
Перспективы людей с наследственной спастической параплегией различаются.
Некоторые люди серьезно страдают и нуждаются в инвалидной коляске, в то время как другие имеют легкие симптомы и не нуждаются в помощи для передвижения.
Заболевание обычно не влияет на продолжительность жизни, и большинство людей могут вести относительно независимый и активный образ жизни.
Национальная служба регистрации врожденных аномалий и редких заболеваний
Если у вас или вашего ребенка есть наследственная спастическая параплегия, ваша клиническая бригада может передать информацию о вас или вашем ребенке в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
NCARDRS помогает ученым определять более эффективные способы лечения или предотвращения редких заболеваний.
Вы можете отказаться от регистрации в любое время.
Узнать больше о регистре NCARDRS
Последняя проверка страницы: 8 августа 2019 г.
Срок следующего рассмотрения: 8 августа 2022 г.
Болезнь Штрюмпеля-Лоррена — История Филиппа Граммона
«Мой отец довольно рано перестал ходить, и ему пришлось передвигаться в инвалидном кресле.Несмотря на это, мы никогда не задумывались о возможности наследственного заболевания в семье », — говорит Филипп Граммон, основатель и вице-президент Французской ассоциации Strümpell-Lorrain (A.SL). «Первые симптомы болезни появились, когда мне исполнилось 35 лет; проблемы при беге и утомляемость в ногах. Три года спустя анализ показал, что мой мозжечок атрофирован. Однако мне поставили правильный диагноз только много лет спустя: болезнь Штрюмпелла-Лоррена, также называемая семейной спастической параплегией.Мой отец был поражен и передал это мне и одному из моих братьев. Два других моих брата и моя сестра не унаследовали дефектный ген. Развитие болезни Штрюмпеля-Лоррена варьируется в зависимости от человека. Вот почему мой брат, близкий мне по возрасту, все еще может ходить с тростью, в то время как мне приходилось использовать инвалидное кресло в течение нескольких лет. Болезнь поразила всю мою семью. Тесты показали, что ген, ответственный за спастическую параплегию, в моем случае является доминирующим, а это означает, что у моих двух дочерей также есть 50% -ный риск заболевания.Они беспокоились об этом много лет, и мы избегали разговоров об этом. В конце концов мы это сделали, и теперь они с нетерпением ждут своего будущего, даже если риск все еще существует ».
В то время, когда у Филиппа появились первые симптомы болезни, наследственная спастическая параплегия была малоизвестна, что создавало у пациентов чувство изоляции. Поэтому в 1992 году Филипп решил создать французскую ассоциацию Strümpell-Lorrain, чтобы объединить французских пациентов. Сегодня ассоциация насчитывает 700 членов на всей территории Франции.Филипп связался со многими пациентами, страдающими спастической параплегией, по всей Европе, чтобы убедить их создать организацию пациентов в своей стране. На сегодняшний день создано 14 организаций, некоторые из них совместно с организациями по атаксии, потому что патологии схожи (проблемы, затрагивающие мозжечок и / или спинной мозг). Филипп даже ездил в Германию, Швейцарию, Англию и Испанию, чтобы развивать европейскую сеть. «Даже если я плохо говорю на других языках, я чувствую себя европейцем, прежде чем почувствовать себя французом», — говорит Филипп.«Чего действительно не хватает сегодня, так это европейской федерации, которая объединила бы все организации под одним знаменем. Иногда пациенты, с которыми я общаюсь, предпочитают создать местное отделение A.SL, а не создавать свою собственную организацию. Это снижает влияние, которое мы могли бы оказать ».
Деятельность Филиппа не принадлежит A.SL. Он играет активную роль в SPATAX 1 , европейской исследовательской сети по атаксии и спастической параплегии, созданной в 2001 году при поддержке INSERM 2 и AFM 3 и объединяющей 27 групп из 13 европейских стран.Сеть собирается раз в год, чтобы поделиться последними результатами исследований. Параллельно со SPATAX Филипп стал участником французской федерации редких заболеваний Alliance Maladies Rares, членом которой является A.SL, и занял должность регионального контактного лица для Franche-Comté (французский регион). Убежденный в важности развития европейских сетей, Филипп обеспечил членство A.SL в EURORDIS. Впоследствии Филипп посетил собрание членов Eurordis в 2003 году в Намюре и в 2006 году в Берлине.«Берлин был для меня уникальной возможностью встретиться с другими людьми, страдающими редкими заболеваниями. Мне не подходит сосредоточение на своей болезни; Мне нужно помочь всем, кто страдает редкими заболеваниями ». А Филипп принял участие в фотоконкурсе Eurordis 2006!
В произвольном порядке некоторые из мероприятий, проводимых Филиппом: организация ежегодной прогулки по Безансону, посвященной редким заболеваниям; организация ежегодного форума по редким заболеваниям во Франш-Конте, который всегда пользуется большим успехом у специалистов в области здравоохранения; создание фильма о спастической параплегии для повышения осведомленности о болезни и поддержки пациентов в их повседневной жизни; и участие в Летнем франкоговорящем университете общественного здравоохранения.Филипп никогда не сдается, даже он жалуется на нехватку волонтеров в этом районе. «Мои проекты помогают мне двигаться вперед, — говорит он. «Занятость другими людьми позволяет мне преодолевать собственные проблемы».
1 Спастическая параплегия
2 Национальный институт здравоохранения и медицинских исследований (Национальный институт здравоохранения и медицинских исследований)
3 Французская ассоциация по борьбе с миопатиями (Французская ассоциация мышечной дистрофии)
Эта статья была ранее опубликована в выпуске нашего информационного бюллетеня за октябрь 2006 г.
Автор: Жером Парисс-Брассенс
Фото предоставлено: © Philippe Grammont / Association Strümpell-Lorrain
Последнее обновление страницы: 13.08.2013
% PDF-1.4 % 708 0 obj> эндобдж xref 708 219 0000000016 00000 н. 0000005631 00000 н. 0000004770 00000 н. 0000005884 00000 н. 0000008876 00000 н. 0000009476 00000 н. 0000009978 00000 н. 0000010014 00000 п. 0000010060 00000 п. 0000010300 00000 п. 0000010546 00000 п. 0000010623 00000 п. 0000011301 00000 п. 0000011927 00000 н. 0000012634 00000 п. 0000013348 00000 п. 0000014766 00000 п. 0000016248 00000 п. 0000017727 00000 п. 0000019217 00000 п. 0000021887 00000 п. 0000021983 00000 п. 0000022118 00000 п. 0000022256 00000 п. 0000022431 00000 п. 0000022606 00000 п. 0000022836 00000 п. 0000023113 00000 п. 0000023348 00000 п. 0000023562 00000 п. 0000023793 00000 п. 0000024030 00000 п. 0000024275 00000 п. 0000024521 00000 п. 0000024766 00000 п. 0000024908 00000 н. 0000025151 00000 п. 0000025293 00000 п. 0000025532 00000 п. 0000025674 00000 п. 0000025809 00000 п. 0000026036 00000 п. 0000026171 00000 п. 0000026399 00000 п. 0000026633 00000 п. 0000026859 00000 п. 0000027090 00000 н. 0000027324 00000 п. 0000027555 00000 п. 0000027763 00000 п. 0000027898 00000 н. 0000028073 00000 п. 0000028215 00000 п. 0000028350 00000 п. 0000028544 00000 п. 0000028686 00000 п. 0000028880 00000 п. 0000029076 00000 п. 0000029270 00000 п. 0000029405 00000 п. 0000029594 00000 п. 0000029736 00000 п. 0000029923 00000 н. 0000030109 00000 п. 0000030305 00000 п. 0000030440 00000 п. 0000030655 00000 п. 0000030868 00000 п. 0000031010 00000 п. 0000031233 00000 п. 0000031371 00000 п. 0000031595 00000 п. 0000031733 00000 п. 0000031958 00000 п. 0000032096 00000 п. 0000032323 00000 п. 0000032461 00000 п. 0000032599 00000 п. 0000032821 00000 п. 0000032959 00000 п. 0000033198 00000 п. 0000033340 00000 п. 0000033564 00000 п. 0000033739 00000 п. 0000033881 00000 п. 0000034106 00000 п. 0000034281 00000 п. 0000034423 00000 п. 0000034647 00000 п. 0000034822 00000 п. 0000034960 00000 п. 0000035102 00000 п. 0000035328 00000 п. 0000035503 00000 п. 0000035735 00000 п. 0000035910 00000 п. 0000036048 00000 п. 0000036286 00000 п. 0000036459 00000 п. 0000036678 00000 п. 0000036848 00000 н. 0000037066 00000 п. 0000037236 00000 п. 0000037374 00000 п. 0000037590 00000 п. 0000037757 00000 п. 0000037895 00000 п. 0000038116 00000 п. 0000038280 00000 п. 0000038418 00000 п. 0000038556 00000 п. 0000038764 00000 п. 0000038924 00000 п. 0000039126 00000 п. 0000039286 00000 п. 0000039424 00000 н. 0000039630 00000 н. 0000039784 00000 п. 0000039922 00000 н. 0000040130 00000 п. 0000040284 00000 п. 0000040490 00000 п. 0000040644 00000 п. 0000040849 00000 п. 0000041000 00000 п. 0000041199 00000 п. 0000041347 00000 п. 0000041485 00000 п. 0000041705 00000 п. 0000041853 00000 п. 0000041991 00000 п. 0000042204 00000 п. 0000042349 00000 п. 0000042487 00000 п. 0000042714 00000 н. 0000042859 00000 п. 0000042997 00000 п. 0000043135 00000 п. 0000043364 00000 п. 0000043502 00000 п. 0000043640 00000 п. 0000043778 00000 п. 0000043990 00000 п. 0000044128 00000 п. 0000044341 00000 п. 0000044479 00000 п. 0000044707 00000 п. 0000044845 00000 п. 0000045076 00000 п. 0000045214 00000 п. 0000045352 00000 п. 0000045552 00000 п. 0000045694 00000 п. 0000045832 00000 п. 0000046032 00000 п. 0000046170 00000 п. 0000046308 00000 п. 0000046508 00000 п. 0000046646 00000 п. 0000046788 00000 п. 0000046988 00000 п. 0000047126 00000 п. 0000047323 00000 п. 0000047461 00000 п. 0000047599 00000 п. 0000047796 00000 п. 0000047938 00000 п. 0000048080 00000 п. 0000048277 00000 п. 0000048474 00000 п. 0000048612 00000 н. 0000048809 00000 п. 0000048947 00000 н. 0000049141 00000 п. 0000049283 00000 п. 0000049421 00000 п. 0000049615 00000 п. 0000049757 00000 п. 0000049899 00000 н. 0000050093 00000 п. 0000050235 00000 п. 0000050426 00000 п. 0000050568 00000 п. 0000050758 00000 п. 0000050953 00000 п. 0000051095 00000 п. 0000051270 00000 п. 0000051412 00000 п. 0000051596 00000 п. 0000051738 00000 п. 0000051880 00000 п. 0000052061 00000 п. 0000052203 00000 п. 0000052378 00000 п. 0000052523 00000 п. Ɔ 䛠 V] = xŽ | iӃ} _
Лечение болезни Бехтерева в клиниках Германии
Теперь можно поехать лечиться в Германию!
Оформляем срочные медицинские визы для въезда в Германию!
Свяжитесь с нами! Мы знаем все о доступных рейсах по всему миру!
Болезнь Бехтерева — хроническое заболевание, вызывающее воспаление межпозвонковых суставов. при развитии анкилоза (неподвижность из-за сращения суставных поверхностей).Это очень распространенная патология. От 0,2 до 2% взрослого населения в разных странах страдают этим заболеванием. Болезнь Бехтерева рано или поздно приведет к инвалидности в результате болей, нарушений подвижности суставов и позвоночника.
На портале Booking Health представлены 69 немецких клиник, специализирующихся на лечении болезни Бехтерева (болезни Мари-Штрампеля)
Показать все клиникиБолезнь Бехтерева — диагностика
Болезнь Бехтерева диагностируется как клинически, так и с помощью Рентгеновские методы.Для установления диагноза необходимо наличие на рентгенограмме таких клинических признаков, как двусторонний сакроилеит в сочетании с 4 из следующих 5 признаков:
- Скованность и болезненные ощущения в крестце, которые не исчезают после отдыха, продолжительностью не менее более 3 месяцев
- Скованность и боль в грудном отделе позвоночника
- Ограничение подвижности в поясничной области
- Ирит (воспаление радужной оболочки) в анамнезе или на момент осмотра
- Ограничение экскурсии грудной клетки при дыхании
Для ранней диагностики болезни Бехтерева используется специальная шкала, в которой суммируются баллы и отдельные симптомы.Учитываются клинические, генетические, лабораторные и рентгенологические признаки.
Лучшие клиники по диагностике болезни Бехтерева в Германии:
Университетская клиника Мюнхенского университета Людвига Максимилиана
0
Узнать большеУниверситетская клиника Шарите Берлин
0
Узнать больше Показать все диагностические программыБолезнь Бехтерева — лечение
Болезнь Бехтерева неизлечима.Более того, в современной медицине нет терапевтических методов, замедляющих прогрессирование болезни. Эта патология плохо поддается терапии. Следовательно, основная цель терапии — поддержание удовлетворительного качества жизни пациента и сохранение его работоспособности. Эта цель прекрасно достигается после лечения в Германии.
Консервативное лечение применяется на ранней стадии болезни Бехтерева. Для пожизненного применения назначают следующие препараты — НПВП, глюкокортикоиды.Для лечения используются лечебная гимнастика, физиотерапия, массаж. Время от времени проводятся курсы плазмафереза или гемосорбции для очистки крови от иммуноглобулинов. Они помогают снять обострения болезни.
В отдельных случаях требуется хирургическое лечение, направленное на восстановление подвижности пациента. При необходимости применяются следующие методики:
- Эндопротезирование сустава (обычно бедра) при анкилозе 3 степени
- Операции на позвоночнике по коррекции угла шеи или поясницы
- Остеосинтез или дорсальная фиксация при переломах шеи
Далее продолжается поддерживающая терапия, направленная на устранение симптомов болезни Бехтерева.Если препараты первого ряда становятся неэффективными, назначают ингибиторы ФНО (фактор некроза опухоли). Лечение в Германии предлагает наиболее рациональное сочетание всех доступных медицинских методик.
Лучшие клиники по лечению болезни Бехтерева в Германии:
Университетская клиника Шарите Берлин
Диагностика и консервативное лечение анкилозирующего спондилита (шейного спондилеза)0
Дорсальная фиксация или остеосинтез у пациентов с переломом шеи и анкилозирующим спондилитом0
Коррекция шейного угла при анкилозирующем спондилите0
Kasselos orthopedic Clinic
Дорсальная фиксация или остеосинтез у пациентов с переломом шеи и анкилозирующим спондилитом0
Коррекция угла шеи при анкилозирующем спондилите0
Показать все программы леченияАвтор: Dr.Надежда Иванисова
В стоимость услуг входит
Здесь Вы можете узнать стоимость лечения этого заболевания в Немецких университетских больницах. Оставьте заявку и мы бесплатно проконсультируем врача и приступим к организации всего лечебного процесса.
В программу входят:
- Оформление приглашения на получение визы на лечение в кратчайшие сроки
- Запись на прием в удобное для Вас время
- Предварительная организация комплексного обследования и обсуждение предстоящего лечения план
- Организация трансфера из аэропорта в больницу и обратно в аэропорт
- Предоставление услуг переводчика и услуг личного медицинского координатора
- При необходимости помощь в организации дальнейшего хирургического лечения
- Предоставление медицинской страховки от осложнения лечения на сумму до 200000 евро
- Подготовка и перевод медицинских карт и рекомендаций из больницы
- Помощь в последующем общении с лечащим врачом, включая консультации по повторным рентгеновским снимкам через уникальная система управления медицинскими документами E-doc
Наследственный спастический парапарез: обзор новых разработок
Наследственный спастический парапарез (HSP) или синдром Стрюмпелла-Лоррена — это название, данное гетерогенной группе наследственных заболеваний, в которых особенность — прогрессирующая спастичность нижних конечностей.До появления молекулярно-генетических исследований этих расстройств было предложено несколько классификаций, основанных на способе наследования, возрасте появления симптомов и наличии или отсутствии дополнительных клинических признаков. Описаны семьи с аутосомно-доминантным, аутосомно-рецессивным и X-сцепленным наследованием.
Исторические аспекты
В 1880 году Струмпель опубликовал то, что считается первым четким описанием HSP. Он сообщил о семье, в которой два брата страдали спастической параплегией.Считалось, что отец был «немного хромым», что предполагало аутосомно-доминантную передачу 1. И Струмпель, и Лоррен добавляли похожие случаи в литературу в последующие годы. 3 Вскоре после этих знаковых описаний в литературе появились многочисленные сообщения о случаях HSP. Однако Пратт, который считал наличие дополнительных неврологических признаков несовместимым с исходными описаниями, назвал многие из них синдромами HSP плюс.4 В ранних обзорах этих и других случаев была сделана попытка идентифицировать «чистые» случаи, как это было описано Струмпеллом. .Однако определение «чистого» варьировалось среди авторов5-8. Именно Хардинг в 1981 году после детальной клинической оценки 22 семей предложил критерии для классификации HSP на чистые и сложные формы, которые с тех пор были приняты и обсуждаются ниже. 9 Дальнейшее подразделение чистого HSP было также предложено Хардингом в зависимости от возраста начала заболевания. Было обнаружено, что семьи можно разделить на две группы: одна с дебютом до 35 лет (тип I), а другая с дебютом после 35 лет (тип II).Казалось, что между группами имелись клинические различия: пациенты типа I имели медленное и вариабельное течение по сравнению с более быстро развивающимся типом II, в котором мышечная слабость, симптомы мочеиспускания и потеря чувствительности были более выраженными.9 Ни одна из этих классификаций не является идеальный, со многими семьями, которые не легко соответствуют критериям. Как и в случае других наследственных нейродегенеративных заболеваний, раскрытие молекулярно-генетических механизмов, лежащих в основе наследственного спастического парапареза, без сомнения, предоставит более полезную и актуальную классификацию.
Эпидемиология
Распространенность HSP варьируется в разных исследованиях. Такие различия, вероятно, связаны с сочетанием различных диагностических критериев, различных эпидемиологических методологий и географических факторов. Некоторые исследования, в которых использовались аналогичные критерии и методы, показали, что распространенность HSP / 100000 составляет 2,7 в Молизе, Италия, 4,3 в Валле-д’Аоста, Италия, и 2,0 в Португалии.10-12 В этих исследованиях использовались диагностические критерии, предложенные Хардингом. и задействовали все медицинские учреждения и различных специалистов в области здравоохранения для установления случаев заболевания в конкретном регионе.Polo и др. в 1991 г. сообщили о более высоком уровне распространенности — 9,6 на 100 000 в Кантабрии, Испания.13 В их отчете для установления случаев использовались только больничные записи, хотя многие вторичные случаи были выявлены путем обследования всех родственников из группы риска.
Клинические особенности
ЧИСТЫЙ НАСЛЕДСТВЕННЫЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ
Критерии, которые были предложены для диагностики чистого HSP, включены в таблицу 1. Большинство пациентов испытывают затруднения при ходьбе или нарушение походки, заметные либо ими самими, либо родственниками.У тех, кто дебютирует в детстве, нередко задержка ходьбы. Другие симптомы включают жесткость ног, нарушение мочеиспускания и преждевременное ношение обуви. До 25% больных протекают бессимптомно, что подчеркивает зачастую доброкачественный характер заболевания и важность тщательной клинической оценки семей, включенных в генетические исследования. Возраст дебюта может быть от младенчества до восьмого десятилетия.9 Заметные межсемейные вариации в возрасте дебюта были одним из первых указателей на генетическую гетерогенность этого состояния.Это изменение также может быть частично связано с трудностями в установлении точной даты начала, особенно у пожилых пациентов, страдающих этим заболеванием в течение десятилетий, или может отражать другие, еще неизвестные генетические или изменяющие окружающую среду факторы.
Таблица 1Предлагаемые диагностические критерии чисто наследственного спастического парапареза и клинические признаки, которые могут предупредить врача о возможных альтернативных диагнозах
Кардинальные отклонения при обследовании пациентов с чистым HSP включают спастичность, гиперрефлексию и подошвенные разгибатели со слабостью пирамидального распределения в нижних конечностях.Спастичность нижних конечностей является заметной находкой при осмотре, особенно в области подколенных сухожилий, четырехглавой мышцы и лодыжек. Этот образец гипертонуса отвечает за классическую походку, при которой пострадавший демонстрирует циркумдукцию и ходьбу на пальцах ног. Слабость мышц, если она присутствует, наблюдается в подвздошно-поясничной, передней большеберцовой мышце и, в меньшей степени, в подколенных сухожилиях. Характерной чертой HSP, которая подчеркивалась несколькими авторами, является заметное несоответствие между часто выраженной спастичностью и только легкой или отсутствующей мышечной слабостью.8 14 Это демонстрирует пациент с HSP, который прикован к инвалидной коляске из-за спастичности, но при ручном мышечном тесте имеет нормальную мощность.
Другие особенности, которые некоторые авторы включили в понятие чистого HSP, включают: легкие сенсорные аномалии в нижних конечностях, отсутствие толчков в голеностопном суставе, кавусную стопу, симптомы мочеиспускания, легкую атаксию в верхних конечностях и умеренное истощение дистальных мышц.9 15-18 Этот более инклюзивный подход, кажется, подтверждается мультисистемным участием, предложенным параклиническими исследованиями и невропатологическими находками.Нарушение чувствительности наблюдается в 10–65% случаев чистого HSP и чаще, но не исключительно, встречается у пациентов с длительным заболеванием. Обычно она заключается в снижении чувствительности к вибрации и, реже, в ослаблении чувства положения суставов в конечностях нижних конечностей. 15 16 У большинства пациентов исследования нервной проводимости в норме. 15 19 20 Это говорит о том, что аномалии, обнаруженные при обследовании, связаны с центральной аксонопатией, а не с поражением периферических нервов. Необычное обнаружение отсутствия рефлексов голеностопного сустава у пациентов с HSP, вероятно, отражает тот же процесс.9 16 Pes cavus — не такая распространенная находка, как предполагалось изначально. Хардинг в своей серии исследований обнаружила кавусную мозоль только у одной трети пациентов.9 Ее наличие может отражать тяжесть или продолжительность заболевания. Нарушение сфинктера мочевого пузыря встречается у 50% пациентов. Обычно это проявляется в сочетании срочности, неуверенности и частоты и чаще встречается при длительном заболевании. 15-17 21 год 22 Вовлечение анального сфинктера необычно для HSP, хотя Schetlens et al действительно сообщили о семье, в которой присутствовали позывы на фекалии и недержание мочи, и Шварц ранее описал мужчину, страдающего аналогичным заболеванием.В обоих отчетах также присутствовали мочевые симптомы. 24 Сексуальная дисфункция описана только в одной семье. Это произошло примерно через 10–15 лет после начала заболевания, поэтому репродуктивная способность пациента не сильно пострадала. Проблема у пораженных мужчин заключалась в эректильной недостаточности с сохранением способности к эякуляции.25 Легкое мышечное истощение хорошо известно, хотя и редко встречается при HSP. Когда он присутствует, он обнаруживается в дистальных мышцах нижних конечностей, обычно в небольших мышцах стопы и передней большеберцовой мышце.Чаще встречается у пациентов, которые болеют этим заболеванием более 10 лет. 15 Если мышечное истощение заметно и присутствует на ранних стадиях заболевания, диагноз следует пересмотреть. Поражение верхних конечностей относительно редко и обычно проявляется легкой гиперрефлексией, которая может присутствовать на ранних стадиях заболевания. След терминальной дисметрии также может быть обнаружен в верхних конечностях, но более ярких признаков мозжечка не наблюдается. Иногда поступают сообщения о легкой атрофии дистальных мышц, но более выраженное поражение верхних конечностей со спастичностью, слабостью или амиотрофией обычно не наблюдается при чистом HSP.9
Важными отрицательными результатами обследования являются нормальная функция черепных нервов и отсутствие признаков поражения кортикобульбарного тракта. Возможное участие вегетативной нервной системы в HSP систематически не изучалось, хотя Картлидж и Боун, сообщая об обнаружении нарушения сфинктера у пациентов с HSP, рассматривали участие вегетативной нервной системы у трех пациентов и выполняли ряд автономных функций. тесты, которые были нормальными.17
Прогноз и тяжесть HSP варьируются между семьями и, в меньшей степени, внутри одной семьи, хотя ожидаемая продолжительность жизни нормальная.Несколько авторов поддержали наблюдение Хардинга о том, что при раннем проявлении HSP (<35 лет) болезнь прогрессирует медленно, большинство пациентов остаются амбулаторными на протяжении большей части своей жизни, и лишь небольшая часть приковывает к инвалидному креслу в пожилом возрасте. Это контрастирует со многими случаями позднего начала HSP (> 35 лет), при которых заболевание может прогрессировать быстро, и большинство пациентов теряют способность ходить в возрасте от 60 до 70 лет.9 16 20
СЛОЖНЫЙ НАСЛЕДСТВЕННЫЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ
В то время как в чистом HSP спастический парапарез является характерной особенностью, в сложном HSP это один из компонентов гораздо более вариабельного фенотипа.Некоторые типы сложных HSP чрезвычайно редки и, возможно, были описаны только в одной семье. В некоторых случаях можно утверждать, что более чем одно наследственное заболевание может влиять на клинические особенности. Наследственный спастический парапарез был связан со многими состояниями, включая атрофию зрительного нерва, ретинопатию, экстрапирамидное заболевание, амиотрофию, деменцию, атаксию, умственную отсталость, глухоту, итиоз, периферическую невропатию и эпилепсию. Ссылки и краткие описания дополнительных клинических признаков приведены в таблице 2.
Таблица 2Клинические особенности, которые могут проявляться помимо спастического парапареза при осложненном наследственном спастическом парапарезе
Эпилепсия, связанная с HSP, не упоминалась в более ранних обзорах, возможно, потому что, имея только редкие сообщения о случаях одного или двух затронутых членов в родословной, связь могла быть случайной, учитывая высокую распространенность эпилепсии в сообществе. Однако за последнее десятилетие несколько авторов опубликовали отчеты о случаях, в которых синдромы эпилепсии и HSP, по-видимому, тесно связаны.39 41-44 Нет четкой закономерности между семьями, в которых существует эта ассоциация. Наблюдается как аутосомно-доминантное, так и рецессивное наследование. Не существует последовательного типа приступов, связанных с HSP, и сообщалось о миоклонической, простой парциальной, сложной парциальной и генерализованной эпилепсии. У пострадавших членов одной семьи иногда описывались различные типы припадков. Точно так же существует вариативность в отношении того, когда приступы начинаются, с примерами припадков до и после начала нарушения походки.В этих семьях часто описывались сопутствующие умственная отсталость или когнитивные нарушения. Эпилепсия является характерным признаком митохондриальных заболеваний (например, MERRF), группы заболеваний, которые часто проявляются различными симптомами, мало чем отличающимися от сложных HSP. Как будет обсуждаться позже, доказательства участия митохондрий были обнаружены в некоторых аутосомно-рецессивных семьях HSP, и будет интересно определить, вносит ли митохондриальная дисфункция вклад в патогенез других форм HSP.
Есть семьи с HSP, у которых, по-видимому, действительно поражены периферические нервы. Шади и Смит сообщили о семье с бессимптомным сенсорным поражением, обнаруженным при клиническом обследовании. У всех членов семьи, которые проходили исследования нервной проводимости, были снижены потенциалы сенсорного действия. Трем пациентам была сделана биопсия нерва, которая показала уменьшение количества миелинизированных волокон большого и среднего размера без признаков демиелинизации, что свидетельствует об аксональной невропатии50. Такие семьи представляют собой примеры сложных HSP.В более ранних сообщениях описывалась тяжелая сенсорная нейропатия, связанная с HSP, с хроническими безболезненными кожными язвами и невропатической резорбцией кости, происходящей в раннем детстве.51 В менее тяжелой форме трофические изменения кожи и язвы на ногах развиваются во взрослой жизни, наложенные на более длительные спастические парапарез.52 53
Деменция была связана с HSP в родословных, где участники могут иметь дополнительные признаки, такие как атаксия, 65 дизартрия, 66 дизартрия и легкая неподвижность лица, 40 сердечные заболевания, 32 эпилепсия, 39 и синдром Маста, который состоит из спастической параплегии с деменцией, дизартрии и т. Д. и атетоз.62 О деменции также сообщалось как в аутосомно-доминантных, так и в рецессивных семьях, как об изолированном сопровождении спастического парапареза. 35–38 В этих семьях были обнаружены сходные модели когнитивных нарушений. К ним относятся различные комбинации дефектной недавней памяти, нарушения внимания, низкой скорости восприятия, плохой зрительно-моторной координации и забывчивости. В описаниях пострадавших не было признаков, указывающих на серьезное поражение коры, таких как дисфазия, агнозия и дискалькулия.Это предполагает, что подкорковая деменция может характеризовать сложный фенотип HSP спастической параплегии с деменцией. Возможно, что более легкие степени когнитивных нарушений могут быть более частыми, чем ранее предполагалось, у людей, страдающих HSP.38 Эта проблема еще не подвергалась систематической оценке. В одном из немногих исследований, в которых тщательно оценивалась когнитивная функция, бессимптомные нарушения были продемонстрированы у пяти из семи пациентов из четырех семей с чистым HSP.67 Таким образом, кажется вероятным, что если пациентов тщательно обследовать на предмет субклинических когнитивных проблем, то даже чистый HSP окажется мультисистемным расстройством с вовлечением вне двигательной системы.Этот феномен все чаще обнаруживается при других явно селективных двигательных расстройствах, включая заболевание двигательных нейронов68. 69
Результаты расследования
Исследования полезны для исключения альтернативных диагнозов, но не повышают достоверность диагностики HSP. Несколько авторов сообщают, что время центральной моторной проводимости показывает либо не регистрируемые, либо отсроченные ответы от нижних конечностей, с обычно нормальными значениями от верхних конечностей.70-73 Сообщается, что соматосенсорные вызванные потенциалы малы или отсутствуют, при этом аномалия снова наблюдается в основном в нижних конечностях. 72 74 Исследования нервной проводимости и ЭМГ являются нормальными в большинстве случаев чистого HSP.9 20 73 75 76
Анализ спинномозговой жидкости при HSP обычно ничем не примечателен. Однако случайные сообщения об аномалиях включали повышение концентрации белка в сложных семьях и повышение концентрации гомокарнозина в сложной семье со спастической параплегией, прогрессирующей умственной недостаточностью и пигментацией сетчатки.18 29 77 78
На МРТ спинной мозг может казаться маленьким, но другие аномалии обычно не видны. Время от времени поступают сообщения о легкой или умеренной атрофии внутричерепных структур, особенно мозолистого тела, а также о поражениях белого вещества в полушариях головного мозга. 71 79 80 Однако заметная атрофия или изменения белого вещества на МРТ требуют тщательного исключения других состояний.
В молекулярной генетике HSP наблюдается стремительный прогресс, который повлияет на то, как мы будем исследовать и диагностировать HSP в будущем, и это будет обсуждаться позже.
Дифференциальная диагностика
Наследственный спастический парапарез следует рассматривать как диагноз исключения. Важно учитывать поддающиеся лечению расстройства, которые могут проявляться аналогичным образом, такие как дефицит B12, допа-зависимая дистония и структурные нарушения спинного мозга. Заболевания, прогноз которых значительно отличается от HSP, такие как рассеянный склероз и семейное заболевание двигательных нейронов, также должны быть исключены. Необходимость исследований будет зависеть от индивидуальной клинической картины.В таблице 3 подробно описаны исследования, которые могут быть полезны для исключения других расстройств у конкретных пациентов. Только после уверенного исключения других диагнозов пациенты и их семьи могут получить соответствующую консультацию.
Таблица 3Дифференциальный диагноз наследственного спастического парапареза
Невропатология
Наследственный спастический парапарез — это заболевание, совместимое с нормальной продолжительностью жизни, при этом большинство пациентов умирают в старости от случайных болезней. Учитывая хронический характер заболевания и ограниченное количество вариантов лечения, значительная часть пациентов может не проходить регулярное обследование у невролога.Поэтому неудивительно, что сообщений о патологии HSP относительно мало. Шварц и Луи проанализировали ранние патологические отчеты в 1952 году, идентифицировали семь случаев чистого HSP и добавили свое собственное описание случаев в 1956 году.81 24 С тех пор было опубликовано лишь несколько патологических отчетов80. 82-84 Основным нейропатологическим признаком чистого HSP является дегенерация аксонов, которая максимальна в конечных частях наиболее длинных нисходящих (кортикоспинальные тракты) и восходящих (пути дорсального столба) трактов спинного мозга.Демиелинизация и глиоз могут сопровождать потерю аксонов. Наиболее сильно поражены проводящие пути — это пересеченные и непересеченные кортикоспинальные пути к нижним конечностям и волокна fasiculus gracilis от нижних конечностей (рисунок). Поражение спиноцеребеллярных трактов наблюдается примерно в 50% случаев. Другие менее распространенные результаты включают уменьшение количества клеток Бец в слое V моторной коры в четырех случаях, заметную потерю нейронов в столбце Кларка в других четырех случаях и один случай, когда мозжечок и базальные ганглии показали потерю нейронов.Ганглии задних корешков, задние корешки и периферические нервы обычно в норме, как и клетки передних рогов, хотя в одном отчете действительно продемонстрирована дегенерация клеток передних рогов по всему спинному мозгу пострадавшего человека.
Срез спинного мозга средней шейки матки из случая сцепленного с хромосомой 2 аутосомно-доминантного чистого HSP (SPG4), окрашенного на миелин Luxol Fast Blue. Имеется диффузная миелиновая бледность пересеченных и непересеченных кортикоспинальных трактов. Кроме того, изящные пучки в центре спинных колонн демонстрируют заметную миелиновую бледность.Клетки переднего рога нормальные по количеству и морфологии.
В единственном количественном исследовании было обнаружено, что размер пирамид у пострадавшего в возрасте 57 лет сопоставим с размером пирамид в среднем у двухлетнего человека. Общее количество миелинизированных нервных волокон в пирамиде составило 376 055 по сравнению с 625 700 у 20-летнего мужчины. В том же исследовании количество клеток Беца было уменьшено до 23 652 в левой моторной области по сравнению с 34 562 у 22-летней женщины. Природа клеточной дегенерации неясна.Уменьшение количества клеток Беца можно объяснить ретроградной дегенерацией, которую можно увидеть, когда повреждение спинного мозга происходит по нескольким механизмам. Учитывая, что дистальная часть длинных аксонов в кортикоспинальных трактах, а также сенсорные и спиноцеребеллярные пути преимущественно затрагиваются в HSP, было высказано предположение, что происходит процесс «отмирания назад», с дегенерацией, начинающейся дистально, а затем продолжающейся по направлению к телу клетки. 0,24
Патологические отчеты иногда выявляли клинически неожиданные проблемы, такие как дегенерация спинного отдела позвоночника у пациентов без обнаруживаемых сенсорных аномалий в течение жизни.В одном случае была задокументирована патология мозжечка и базальных ганглиев при аутопсии без каких-либо задокументированных клинических коррелятов.
Недавно были описаны новые патологические изменения в случае чистого HSP с аутосомно-доминантным наследованием, сцепленного с локусом хромосомы 2. Описанный пациент имел ранний анамнез, полностью совместимый с чистым HSP, у него развивался прогрессирующий спастический парапарез в возрасте 19 лет. Однако в возрасте 72 лет у него развилось быстро прогрессирующее дементирующее заболевание с несколькими эпилептическими припадками на претерминальных стадиях.Патология спинного мозга полностью соответствовала чистому HSP. Однако патология головного мозга была необычной. Интересные особенности включали потерю нейронов в черной субстанции с ассоциированными тельцами Леви и бледными тельцами, а также изменения гиппокампа с тау-иммунореактивными нейрофибриллярными клубками. Другой примечательной особенностью было наличие тау-положительных телец включения в лимбической области и неокортексе. Эти изменения ранее не наблюдались при HSP и также не соответствуют патологии, характерной для других определенных форм деменции.Поэтому возможно, что эти церебральные патологические особенности являются прямым результатом экспрессии аномального продукта гена SPG440.
Генетика
Клиническая гетерогенность HSP сочетается с генетической гетерогенностью. Аутосомно-доминантные, рецессивные и X-сцепленные семьи хорошо распознаются с несколькими описанными локусами. Краткое изложение текущей генетической классификации HSP показано в таблице 4.
Таблица 4Современная генетическая классификация наследственного спастического парапареза
Аутосомно-доминантный HSP (ADHSP)
Аутосомно-доминантное наследование является наиболее распространенным типом наследования при HSP и связано с локусами на хромосоме 2p (SPG4), 38-40 71 86-94 хромосома 14q (SPG3), 88-90 95-97 хромосома 15q (SPG6), 98 хромосома 8q (SPG8), 99 хромосома 10q (SPG9), 64 и хромосома 12q (SPG10).100
ХРОМОСОМ 2P LOCUS SPG4
Локус хромосомы 2 (SPG4) на сегодняшний день является наиболее распространенным локусом, с которым связаны семейства ADHSP, составляя около 40% родословных. Недавно ген, ответственный за HSP в локусе SPG4, был идентифицирован с использованием стратегии позиционного клонирования. Ген спастин расположен на хромосоме 2p22-p21 и состоит из 17 экзонов, охватывающих область размером 90 т.п.н. 101. Спастин является членом группы белков, известных как АТФазы, связанные с различными клеточными активностями (AAA).Белки ААА участвуют в различных клеточных функциях, включая регуляцию клеточного цикла, деградацию белков, биогенез органелл и функцию белка, опосредованную пузырьками.102 Все эти клеточные механизмы включают сборку и функцию белковых комплексов, и было высказано предположение, что действуют члены семейства ААА. как белки-шапероны в этих комплексах.103 Белки AAA имеют общий домен из 230 аминокислот, который обеспечивает функцию АТФазы. Вне этого домена наблюдается очень небольшая гомология, за исключением близкородственных белков того же подсемейства белков AAA.102 Интересно, что параплегин, член другой подгруппы белков AAA, как было показано, участвует в аутосомно-рецессивном HSP SPG7 (см. Ниже) .104 Спастин имеет небольшую гомологию с параплегином, за исключением домена AAA и, в отличие от параплегина, предположительно имеет ядерная локализация. Спастин демонстрирует большую гомологию с конкретным подклассом белков ААА, которые включают субъединицы 26S протеасомы дрожжей.101 Было высказано предположение, что ядерные белки ААА, которые проявляют гомологию с субъединицами 26S протеасомы, играют косвенную роль в регуляции генов, вызывая протеолитическую активацию или деградацию факторов транскрипции. .105
На сегодняшний день идентифицированные мутации спастина включают бессмысленные, бессмысленные и сплайсинговые мутации в различных экзонах, приводящие к серьезным изменениям в аминокислотной последовательности домена AAA. Это предполагает, что SPG4 вызывается потерей функции, что означает, что пороговый уровень экспрессии белка спастина имеет решающее значение для сохранения аксонов в кортикоспинальном тракте.101
Хотя большинство родословных SPG4 имеют чистый HSP, было три семейства со сложными фенотипами, также связанными с этим локусом.38-40 В семьях чистых HSP, связанных с этим локусом, наблюдаются выраженные межсемейные и внутрисемейные вариации по степени тяжести и наличию или отсутствию таких особенностей, как симптомы со стороны мочевого пузыря и снижение чувствительности к вибрации.
Не все опубликованные исследования комментируют серьезность двигательной дисфункции у пострадавших людей, а у тех, кто часто это делает, непросто напрямую сравнивать различные отчеты. Существует тенденция к разной степени тяжести, при которой большинство пострадавших людей сохраняют способность ходить самостоятельно или с минимальной поддержкой.В крайнем случае — бессимптомные пациенты с пирамидными признаками в нижних конечностях с нормальной или лишь слегка ненормальной походкой и несколько пациентов, прикованных к креслу или прикованных к постели71. 94
В семьях, сцепленных с хромосомой 2p, наблюдается заметная вариабельность возраста начала симптомов. Несколько опубликованных родословных показывают возраст появления симптомов в пределах 5 или более десятилетий у разных членов одной семьи. Средний возраст появления симптомов из опубликованных родословных колеблется от 12 до 43 лет, что также свидетельствует о межсемейных вариациях.Причина такой межсемейной и внутрисемейной изменчивости неизвестна. Он может представлять аллельную гетерогенность или присутствие модифицирующих генов или факторов окружающей среды. Другим фактором может быть очевидная сложность определения точного времени появления симптомов, которые начинаются незаметно и часто присутствуют в течение многих лет.
В некоторых семьях сообщается, что в последующих поколениях возраст начала заболевания снижается40. 71 89 92 Это наблюдается при других нейродегенеративных заболеваниях, например, болезни Хантингтона, зубочелюстно-паллидолизийской атрофии (DRPLA) и болезни Мачадо-Джозефа (MJD).Однако эти заболевания вызваны аномальной экспансией повторов CAG, и именно увеличение длины экспансии повторов коррелирует с более ранним началом заболевания в последующих поколениях.106-108 До открытия гена спастина велись обширные поиски доказательства того, что SPG4 был нарушением тринуклеотидных повторов. Как клинические, так и молекулярно-генетические доказательства того, являются ли расширенные повторы CAG патогенными в SPG4, неубедительны.109-111 Однако ген спастина не содержит аномально увеличенных повторов CAG, и должно быть альтернативное объяснение явного ожидания, наблюдаемого в некоторых семьях SPG4. искать.Это может быть просто так, когда сообщается об ожидании, это может фактически отражать предвзятость установления, при которой заболевание выявляется в раннем возрасте у детей пострадавших родителей.
Интересно, что в трех семьях, сцепленных с хромосомой 2p, HSP был связан с когнитивными нарушениями. Уэбб и др. описали большую семью с 12 больными членами. Один пациент умер от слабоумного заболевания в возрасте 62 лет, а у четырех членов семьи с HSP при нейропсихологическом тестировании было обнаружено слабоумие подкоркового типа.38 Во французской семье, описанной Heinzleff et al , четверо из семи затронутых членов семьи имели когнитивные нарушения, тогда как один имел тяжелую деменцию с некоторыми корковыми особенностями. Трое затронутых членов с когнитивными нарушениями также страдали эпилепсией.39 В семье, о которой сообщили White et al , член индекса с HSP умер с поздним началом тяжелого дементирующего заболевания с корковыми и экстрапирамидными особенностями. Два других члена семьи имели нарушение памяти в пожилом возрасте, хотя подробная информация о двигательных симптомах у этих членов отсутствовала.Еще один член семьи с HSP имел пограничные трудности в обучении с легкими нарушениями всех аспектов памяти. Как и в других семьях, были члены семьи с HSP без когнитивных изменений. Патологические находки у пациента с деменцией в этой родословной уже обсуждались ранее40.
ХРОМОСОМ 14Q LOCUS SPG3
Первым обнаруженным локусом ADHSP был SPG 3 на хромосоме 14q, и только пять опубликованных семейств были сцеплены с этим локусом. Расположение гена SPG3 на хромосоме 14q11.2-q24,3 сузился до области 7 см. 88 90 95–97 Среди пяти семей действительно наблюдается тенденция к относительно раннему возрасту начала заболевания, при этом среднее значение обычно находится в пределах первого или второго десятилетия. Не было никаких доказательств, позволяющих предположить, что в последующих поколениях предвосхищение возраста наступает. Первоначально считалось, что фенотип SPG 3 также может быть более серьезным, чем другие подтипы HSP. Однако Хуанг и др. и Гисперт и др. опубликовали родословные, в которых доля серьезно пораженных больных (нуждающихся в инвалидной коляске) не отличается от той, что наблюдается в некоторых родословных, связанных с SPG4.
По сравнению с другими локусами, о которых упоминалось, в фенотипе SPG 3, по-видимому, отсутствует участие мочевого пузыря или органов чувств.
ХРОМОСОМА 15Q LOCUS SPG6
Только одно большое семейство из Северной Америки было связано с SPG6. Локус был сопоставлен с интервалом ~ 7,3 сМ в центромерной области хромосомы 15q.98 Средний возраст начала заболевания составлял 22 года (12–35), и в целом фенотип был чистым HSP, хотя фенотип относительно тяжелый. .Почти треть пострадавших членов нуждалась в инвалидной коляске, некоторым из них было уже 40 лет. Pes cavus почти всегда присутствовала у пораженных членов, что, возможно, отражает тяжесть заболевания.
ХРОМОСОМА 8Q ЛОКУС (SPG8)
Две семьи были сцеплены с локусом SPG8 на хромосоме 8q24. Одна большая североамериканская семья, а в последнее время и британская семья, позволили уточнить генетический локус до области 3,4 сМ.99 112 Средний возраст появления симптомов в родословных, связанных с SPG8, был сопоставим с семьями SPG4.Фенотип казался довольно серьезным, особенно в американской семье, в которой 10 из 15 больных членов семьи старше 40 лет требовали использования инвалидной коляски, причем восемь из них более 50% времени.113 В обеих родословных клинические особенности были типичными для чистого HSP с некоторыми членами семьи, имеющими снижение чувствительности к вибрации, симптомов со стороны мочевого пузыря и кавуса. Магнитно-резонансная томография показала выраженную атрофию спинного мозга у одного пациента с умеренными симптомами из американской семьи, связанной с хромосомой 8q.Было обнаружено уменьшение площади поперечного сечения на 50% на уровне Т9 по сравнению с контрольной группой того же возраста и пола113.
Доказательства митохондриальной дисфункции как механизма заболевания в этом локусе не были обнаружены при гистохимическом или биохимическом анализе мышечной биопсии, взятой у одного пораженного члена113.
ХРОМОСОМ 10Q LOCUS SPG9
Одна крупная итальянская родословная была связана с локусом SPG9 на хромосоме 10q23.3–24.2 в регионе, охватывающем 12 сМ.64 Средний возраст появления симптомов варьируется от первого до третьего десятилетия жизни, и авторы предлагают доказательства для ожидания на протяжении трех поколений.Клинический фенотип — один из сложных HSP со спастическим парапарезом, связанным с двусторонней катарактой, гастроэзофагеальным рефлюксом с постоянной рвотой и дистальной амиотрофией, вторичной по отношению к явной аксональной моторной невропатии. Родословная со схожим сложным фенотипом врожденной катаракты и спастического парапареза была описана ранее, что также может быть связано с тем же геном в локусе SPG9114.
ХРОМОСОМА 12Q LOCUS SPG10
Последним обнаруженным локусом ADHSP является SPG10 на хромосоме 12q13, охватывающий 9.2 сМ в британской семье100. Средний возраст появления симптомов у пораженных членов семьи был моложе, чем в родословных SPG4, и составлял 10,8 (SD 9,6) лет. Фенотип был одним из чистых HSP с широким диапазоном тяжести заболевания: некоторым пациентам требовалась инвалидная коляска, тогда как другие оставались бессимптомными в возрасте 40 лет.
Reid et al описали еще одну родословную, в которой было исключено сцепление со всеми пятью известными локусами ADHSP, что предполагает наличие по крайней мере еще одного, еще не открытого локуса ADHSP.100
Аутосомно-рецессивный HSP (ARHSP)
Рецессивное заболевание встречается гораздо реже, чем доминирующая форма. Видны как чистые, так и сложные фенотипы. Есть кровные семьи, связанные с тремя локусами: SPG5 на хромосоме 8p, 115SPG7 на хромосоме 16q116 и недавно обнаруженный локус на хромосоме 15q, 117, который в настоящее время не имеет официального обозначения в базе данных генома.
ХРОМОСОМА 8P LOCUS SPG5
Локус SPG5 находится на хромосоме 8p12-q13 и охватывает интервал 32 см.Четыре тунисские семьи, связанные с SPG5, имеют чистый фенотип со средним возрастом начала заболевания от 1 до 20 лет. Кажется, что нет разницы между клиническими характеристиками чистого ADHSP и чистого ARHSP.115
ХРОМОСОМА 16Q LOCUS SPG7
Локус SPG7 находится на хромосоме 16q24.3, и в настоящее время с этим локусом сцеплены три семейства. Фенотип неоднороден, встречаются как чистые, так и сложные семейства. Исходное семейство, связанное с SPG7, было описано как чистое, хотя были дополнительные особенности, обычно не наблюдаемые в чистом HSP, присутствующие в этом семействе, о которых сообщали DeMichele et al , которые включали дизартрию.116
Белок, кодируемый SPG7, получил название параплегин. Недавно в этом исходном семействе, сцепленном с SPG7, была описана гомозиготная делеция 9,5 т.п.н. в последних пяти экзонах гена параплегина на хромосоме 16q. Когда другие неродственные пациенты с HSP были подвергнуты скринингу с использованием анализа одноцепочечного подтверждающего полиморфизма (SSCP), были выявлены две дополнительные мутации сдвига рамки считывания в гене параплегина. Один произошел в семье чистых ARHSP из Италии, у пораженных членов обнаружена делеция 2 п.н. в экзоне 6.Другой был обнаружен в сложной семье ARHSP, дополнительные клинические признаки которой включали атрофию зрительного нерва с атрофией коры головного мозга и мозжечка, видимой на изображениях. Эта мутация состояла из вставки в кодирующую область последнего экзона. Все три мутации SPG7 приводят к серьезным нарушениям белка параплегина. Параплегин является членом митохондриальных металлопротеиназ, которые являются подгруппой семейства белков AAA. Другие члены этой подгруппы включают митохондриальные белки дрожжей Afg3p и Rcalp, которые проявляют как протеолитическую, так и шапероноподобную активность и локализуются на внутренней мембране митохондрий.Исследования нацеливания и локализации показали, что белок параплегина локализуется в субклеточном компартменте митохондрий. Обнаружение доказательств митохондриальной дисфункции в мышечной ткани при биопсии у затронутых членов этих семей подтверждает участие митохондрий в процессе болезни. Мышечные аномалии заключались в наличии волокон, отрицательных по цитохромоксидазе, и рваных красных волокон, которые демонстрировали интенсивное окрашивание на сукцинатдегидрогеназу, изменения, которые указывают на нарушение митохондриального OXPHOS.104 Биопсия мышцы пациента с ARHSP, сцепленным с хромосомой 8q, не выявила доказательств митохондриальной дисфункции, что позволяет предположить альтернативный механизм заболевания при этом типе HSP113.
Митохондриальная дисфункция была выявлена при некоторых нейродегенеративных заболеваниях. При таких заболеваниях, как болезнь Паркинсона, может быть прямое участие комплексов дыхательной цепи. При атаксии Фридрейха и болезни Хантингтона дефекты OXPHOS кажутся вторичными по отношению к изменениям белков, отличных от ферментов дыхательной цепи.118
Недавно ген AFG3L2, родственный параплегину, был идентифицирован на хромосоме 18p11.119 Предсказанный генный продукт имеет гомологию с параплегином, митохондриальными АТФазами дрожжей, Afg3p и Rcalp и родственным геном AFG3L1119. 120 AFG3L2 также локализуется в субклеточном компартменте митохондрий. Требуются дальнейшие исследования, чтобы установить, насколько распространены дефекты параплегина в различных формах HSP, и установить, участвует ли AFG3L1 или AFG3L2 в возникновении HSP, особенно в семьях со сложным фенотипом, где мультисистемное вовлечение может напоминать клинические признаки, обнаруженные в другие митохондриальные нарушения.
Этот класс гена может быть вовлечен в другие неврологические заболевания, и интересно, что AFG3L2 находится в области хромосомы 18p, с которой связана форма идиопатической торсионной дистонии121.
ХРОМОСОМ 15Q
После открытия SPG5 и SPG7 все еще оставались семейства ARHSP, не связанные ни с какими локусами. Murrilo и др. недавно обнаружили то, что в настоящее время, по-видимому, является наиболее распространенным локусом для ARHSP на хромосоме 15q13-15.117. Они проверили восемь семей и обнаружили доказательства сцепления с этим новым локусом в семи.
Фенотип в этих семьях вариабелен, описываются как чистые, так и сложные формы. В чистых семьях возраст начала заболевания был в первом десятилетии в одной и в возрасте 43 лет в другой, в которой был затронут только один член семьи. Между сложными семьями также наблюдались фенотипические различия, хотя некоторые сходные черты присутствовали в более чем одной семье. Наиболее интересной была атрофия или агенезия мозолистого тела, продемонстрированная на МРТ, наблюдаемая как у пораженных братьев и сестер из одной семьи, так и у одного пораженного члена из другой.У этого последнего члена также были обнаружены перивентрикулярные изменения белого вещества. Обе эти семьи имели одинаковый возраст появления симптомов болезни во втором десятилетии жизни, и умственная отсталость была характерной чертой, отмеченной у одного члена от каждой семьи. Другими ассоциациями со спастической параплегией в этом локусе были смешанная моторная и сенсорная нейропатия в одной семье и легкая дисфункция мозжечка в двух других.
Связь HSP, связанного с хромосомой 15q, с агенезом мозолистого тела и умственной отсталостью представляет интерес, потому что было показано, что ген синдрома Андермана или агенеза мозолистого тела и периферической невропатии (ACCPN) также связан с хромосомой 15q.122 Возможно, эта форма HSP и синдром Андермана являются аллельными нарушениями с разными мутациями в одном и том же гене, приводящими к различным фенотипам.117 Это наблюдается в X-сцепленном HSP, где разные мутации в гене PLP приводят к возникновению HSP фенотип или болезнь Пелицея-Мерцбахера.
Х-сцепленный HSP
Хотя Х-сцепленный HSP встречается редко, его молекулярно-генетическая основа относительно хорошо изучена.
Существует два разных локуса, идентифицированных для X-сцепленного HSP, SPG1 и SPG2.60 123 Семьи со сложным HSP были связаны как с SPG1, так и с SPG2, тогда как чистый фенотип был связан только с SPG2.
X-LINKED SPG2 LOCUS
Осложненный HSP, связанный с SPG2, состоит из основного фенотипа спастического парапареза, мозжечкового синдрома и умственной отсталости. Наблюдается межсемейная изменчивость, при которой основные признаки проявляются в различной степени с дополнительными признаками, такими как атрофия зрительного нерва, или без них. Аналогичным образом происходит внутрисемейная изменчивость124. 125 Об этом сообщалось в более ранних описаниях X-сцепленных семей.126
Ген в локусе SPG2 представляет собой ген протеолипидного белка (PLP). Известно, что мутации, чаще всего дупликации, в этом гене вызывают болезнь Пелицея-Мерцбахера (ПМД). Это дисмиелинизирующее заболевание, поражающее ЦНС, которое состоит из ранних мозжечковых и пирамидных признаков с быстрым прогрессированием, приводящим к смерти, как правило, в младенчестве или детстве. Было показано, что сложные родословные HSP, сцепленные с локусом SPG2, имеют мутации в гене PLP в экзонах 3b, 4 или 6.127 128 Следовательно, разные мутации в одном и том же гене могут вызывать либо фенотип PMD, либо сложный фенотип HSP. PLP является основным белком миелина, участвующим как в созревании миелина, так и в поддержании его в ЦНС. Менее тяжелый фенотип и более позднее развитие признаков у SPG2 по сравнению с PMD связано с тем, что PLP имеет меньшую изоформу, DM-20. И в SPG2, и в PMD концентрации PLP снижены, тогда как концентрация DM-20 значительно снижается только в PMD. Считается, что эти два изомера немного по-разному влияют на миелин.Изоформа DM-20, по-видимому, преимущественно участвует в созревании миелина, тогда как изоформа PLP, которая экспрессируется на более поздней стадии развития, играет роль в уплотнении и поддержании миелинового листа. Миелиновые оболочки, в которых отсутствует белок PLP, будут собираться, и хотя они могут функционировать, они в конечном итоге претерпевают ускоренный оборот.129 Эти различия могут объяснять заметные фенотипические вариации между SPG2 и PMD.
Было обнаружено еще одно сложное семейство, связанное с областью, содержащей локус SPG2, без мутации, идентифицированной в гене PLP.Авторы предполагают, что это может свидетельствовать о близлежащем третьем локусе для X-сцепленного HSP.130 Однако у части пациентов с ПМД мутации не выявлены.131 132 Это может быть связано с наличием нераспознанных мутаций в некодирующих областях гена или с мутацией в соседнем гене, участвующем либо в регуляции гена PLP, либо в образовании миелина.
Чистый X-связанный HSP очень редок, на сегодняшний день в литературе описано только пять семейств.123 133-136 Два из этих семейств были сцеплены с локусом SPG2.123 133 Только в одном из этих семейств была обнаружена замена единственного основания в экзоне 5 гена PLP, показывая, что родословная чистого HSP является аллельной по отношению к сложному HSP (SPG2) и PMD.
X-LINKED LOCUS SPG1
Семьи с осложненными БТШАМИ связана с SPG1 пометили фенотипические различия по сравнению с предыдущими родословными, в числе которых отсутствие мозжечка участия, тяжелой умственной отсталость, а также врожденные аномалии опорно-двигательный аппарат, в первую очередь отсутствие разгибателей сгибателя.60 Этот фенотип обусловлен мутацией в экзоне 26 гена молекулы адгезии клеток L1 (L1CAM) 61. L1CAM представляет собой трансмембранный гликопротеин, который в основном экспрессируется нейронами и шванновскими клетками. Он играет важную роль в развитии нервной системы, участвуя в росте аксонов и поиске пути. Различные мутации в этом гене вызывают синдромы MASA (умственная отсталость, афазия, шаркающая походка и приведение больших пальцев), Х-сцепленная гидроцефалия и Х-сцепленная агенезия мозолистого тела.В семьях с мутациями в гене L1CAM существует большая межсемейная и внутрисемейная изменчивость. В одной семье могут быть члены с различными фенотипами. Заболевания теперь называют частью клинического синдрома CRASH (гипоплазия мозолистого тела, задержка развития, приведение больших пальцев, спастическая параплегия и гидроцефалия) .137
Заключение
Неудивительно, что на раннем этапе возникли трудности при попытке классифицировать HSP, который представляет собой гетерогенную группу заболеваний, которые в лучшем случае могут иметь общий конечный путь в дегенеративном процессе нейронов.Молекулярно-генетическая классификация сейчас начинает определять пределы условий и их корреляции генотип / фенотип.
В настоящее время мутации в четырех генах; L1CAM, PLP, параплегин и спастин были идентифицированы как играющие роль в развитии фенотипа HSP, при этом идентичность генов в других семи локусах все еще неизвестна. Недавнее открытие генов спастина и параплегина открывает новую эру захватывающих возможностей, в которой мы можем начать исследовать физиологию двигательной системы и дегенеративный процесс, которому она подвергается, что, как мы надеемся, открывает возможности для лечения.В ближайшем будущем можно ожидать открытия остальных генов и их продуктов, что поможет нам лучше понять эти заболевания.
Наличие такой генетической гетерогенности, лежащей в основе HSP, некоторым образом объясняет заметные фенотипические вариации, наблюдаемые между семьями. Однако обнаружение сходных внутрисемейных вариаций можно объяснить только наличием генетических модифицирующих факторов или неизвестным взаимодействием с окружающей средой. Meyer и др. продемонстрировали мутации в гене транспортера глутамата EAAT2 у двух из семи членов семьи, пораженных чистым HSP.Они постулируют, что взаимодействие с аллельным вариантом EAAT2 и неизвестным первичным генетическим дефектом может быть ответственным за фенотипические вариации, наблюдаемые в их семье138.
Несмотря на прогресс в генетике HSP, до сих пор его диагностическая значимость была относительно небольшой. Теперь это может измениться с открытием гена спастина, мутации в котором могут составлять до 50% ADHSP. При осмотре пациента с возможным HSP первым шагом остается исключить другие диагнозы.Генетический анализ даст возможность поставить точный диагноз в случаях выявления мутаций. В конечном итоге это даст возможность предсимптомной или даже пренатальной диагностики в семьях, которые хотят пройти такое обследование. Однако до тех пор, пока не будет понятен полный диапазон мутаций этих генов, техническая возможность проведения такого анализа остается неясной. Это мультиэксонные гены с различными мутациями, поэтому идентификация мутации может быть не очень простой.Тем не менее, возможность точного диагноза знаменует новую область лечения в HSP. В ближайшие годы можно ожидать дальнейшего быстрого прогресса как в идентификации новых генов, связанных с HSP, так и в клеточной биологии, лежащей в основе дегенерации двигательной системы.
Благодарности
PJS поддерживается Wellcome Trust в качестве старшего научного сотрудника в области клинических наук.
Анкилозирующий спондилит | Сидарс-Синай
Не то, что вы ищете?Что такое анкилозирующий спондилит?
Анкилозирующий спондилит (АС) — это тип артрита, поражающего позвоночник.Анкилозинг означает жесткий или жесткий. Спондил означает позвоночник. Это относится к воспалению. Болезнь вызывает воспаление позвоночника и крупных суставов, в результате чего возникает скованность и боль. В болезнь может повредить сустав между позвоночником и тазовой костью. Это называется крестцово-подвздошный сустав. Это также может привести к образованию костных мостов между позвонками в позвоночник, сплавление этих костей. Кости в груди также могут срастаться.
Что вызывает анкилозирующий спондилит?
Врачи не знают, что вызывает AS, но исследователи считают, что гены играют роль. А ген под названием HLA-B27 встречается у большинства кавказских американцев с AS. Но только в половине Афроамериканцы, у которых это есть. Но у некоторых людей с геном HLA-B27 нет АС. Меньше чем 1 из 20 человек с геном HLA-B27 имеет AS.
Кто подвержен риску заболевания анкилозирующим спондилитом?
AS чаще встречается у людей в возрасте от 17 до 35 лет. Это может произойти у детей и старше. взрослые тоже. Заболевание чаще поражает молодых мужчин, чем женщин. Это имеет тенденцию вбегать семьи.
Каковы симптомы анкилозирующего спондилита?
Симптомы АС имеют тенденцию появляться и исчезать со временем.Симптомы могут проявляться немного по-другому в каждый человек. Симптомы могут включать:
- Боль в спине, обычно наиболее сильная ночью в покое
- Рано утром скованность
- Наклонная осанка в ответ на боль в спине (наклон вперед, как правило, облегчает боль)
- Позвоночник прямой и жесткий
- Неспособность сделать глубокий вдох при поражении суставов между ребрами и позвоночником
- Потеря аппетита
- Похудание
- Усталость
- Лихорадка
- Анемия
- Боль в суставах
- Легкое воспаление глаз
- Повреждение органов, например сердца, легких и глаз
- Сыпь на коже
- Заболевание органов пищеварения (например, болезнь Крона или язвенный колит)
Симптомы анкилозирующего спондилита могут быть похожи на другие состояния здоровья.Убеждаться к обратитесь к своему врачу для постановки диагноза.
Как диагностируется анкилозирующий спондилит?
Диагностика начинается с истории болезни и физического осмотра. Также могут понадобиться тесты, такой в виде:
- Рентген. В этом тесте используется небольшое количество излучения для создания изображений внутренних тканей, кости и органы на пленку.
- Эритроцит скорость седиментации (СОЭ или скорость седиментации). Этот тест показывает, насколько быстро красный кровяные тельца падают на дно пробирки. Когда отек и воспаление В настоящее время белки крови слипаются и становятся тяжелее, чем обычно. Они падают и быстрее оседают на дне пробирки. Чем быстрее клетки крови падать, тем сильнее воспаление.До 7 из 10 человек с СА имеют высокий СОЭ.
- Генетический тестирование. Генетическое тестирование проводится, чтобы определить, есть ли у человека копия измененный ген болезни. Ген HLA-B27 обнаружен более чем у 19 из 20 человек. с AS.
Как лечится анкилозирующий спондилит?
Лечение будет зависеть от ваших симптомов, вашего возраста и общего состояния здоровья.Так и будет также зависят от того, насколько тяжелое состояние. Цель лечения — облегчить боль и жесткость, предотвращение деформаций и поддержание как можно более нормального образа жизни. Лечение может включать:
- Нестероидные противовоспалительные препараты (НПВП) для облегчения боли и воспаление
- Блокаторы фактора некроза опухолей (биологические препараты) для облегчения воспаления и припухлость
- Ингибиторы интерлейкина-17A (IL-17A) для облегчения воспаления и отека
- Модифицирующие болезнь противоревматические препараты (DMARD), такие как сульфасалазин, для облегчения воспаление и контроль АС
- Кратковременное применение кортикостероидов для облегчения воспаления
- Кратковременное применение миорелаксантов и болеутоляющих средств для облегчения сильной боли и спазмы мышц
- Операция по замене сустава, установке стержней в позвоночник или удалению утолщенных частей и закаленная кость
- Поддержание правильной осанки
- Упражнения регулярные, в том числе упражнения на укрепление мышц спины
Поговорите со своим лечащим врачом о рисках, преимуществах и возможных побочных эффектах. всех лекарств.
Какие возможны осложнения анкилозирующего спондилита?
Больше Со временем у людей с АС может развиться прямой изгиб позвоночника. Люди с AS в большой риск истончения костей (остеопороз). Это может привести к переломам позвоночника. АС также может привести к псориазу и воспалению глаз, аортального клапана и кишечника. тракт.
Жизнь с анкилозирующим спондилитом
От AS нет лекарства, поэтому важно разработать план лечения вместе с вашим врачом. поставщик медицинских услуг. Работайте над изменениями образа жизни, которые могут улучшить качество вашей жизни. Оставайтесь активными и выполняйте упражнения, чтобы уменьшить боль. Физиотерапевт может помочь ты составьте план упражнений и помогите сохранить хорошую осанку.
Когда мне следует позвонить своему врачу?
Если ваши симптомы ухудшаются или у вас появляются новые симптомы, сообщите об этом своему лечащему врачу. знать.
Основные сведения об анкилозирующем спондилите
- Анкилозирующий спондилит — это разновидность артрита, поражающего позвоночник.
- Ген может быть частью причины AS.
- Симптомы АС включают боль в спине, скованность по утрам и сутулость.
- AS может вызывают другие симптомы, такие как потеря аппетита, потеря веса, усталость, лихорадка, анемия, глаз воспаление и болезни пищеварения.
- Цель лечения АС заключается в облегчении боли и скованности, предотвращении деформаций и поддержании как можно более нормальный образ жизни.
- В управлении AS важно оставаться активным.
Следующие шаги
Советы, которые помогут вам получить максимальную пользу от визита к врачу:
- Знайте причину вашего визита и то, что вы хотите.
- Перед визитом запишите вопросы, на которые хотите получить ответы.
- Возьмите с собой кого-нибудь, кто поможет вам задать вопросы и запомнить, что говорит ваш поставщик ты.
- Во время визита запишите название нового диагноза и любые новые лекарства, методы лечения, или тесты. Также запишите все новые инструкции, которые дает вам ваш провайдер.
- Узнайте, почему прописано новое лекарство или лечение и как они вам помогут. Также знать, каковы побочные эффекты.
- Спросите, можно ли вылечить ваше состояние другими способами.
- Знайте, почему рекомендуется тест или процедура и что могут означать результаты.
- Знайте, чего ожидать, если вы не примете лекарство, не пройдете тест или процедуру.
- Если у вас назначена повторная встреча, запишите дату, время и цель для этого посещение.
- Знайте, как вы можете связаться с вашим провайдером, если у вас есть вопросы.