причины, симптомы, диагностика, лечение, профилактика
Представляет собой патологическое выпячивание в области подколенной ямки, которое образуется на фоне воспалительного поражения коленного сустава.
ПричиныПрактически у каждого второго человека в области задней поверхности коленного сустава, между сухожилиями полуперепончатой и икроножной мышц находится слизистая межсухожильная сумка, наличие которой считается вариантом нормального анатомического строения. При воспалительном поражении коленного сустава происходит скопление жидкости, которая через тонкие щели может проникать в межсухожильную сумку. В следствии таких процессов сумка увеличивается в размерах, появляются боли и ограничение движений в коленном суставе. Подколенная киста Беккера может формироваться в результате обменно-дистрофических и воспалительных заболеваниях.
СимптомыНа начальной стадии заболевание протекает бессимптомно либо сопровождается возникновением незначительных неприятных ощущений. При дальнейшем увеличении в размерах киста начинает сдавливать расположенные поблизости нервы, что становится причиной развития болей, покалывания или онемения в области подошвы, а также не позволяет пациенту полностью сгибать коленный сустав.
При фтизикальном осмотре у таких больных иногда выявляется видимое опухолевидное образование в подколенной ямке. При пальпации киста Беккера определяется незначительная болезненность. На ощупь образование кажется упругим и плотным.
Основным осложнением кисты Беккера является ее разрыв, во время которого жидкость из суставной сумки поступает в мышцы, что становится причиной появления болей и отека в области задней поверхности голени.
Иногда данное патологическое образование может вызывать сдавление вен голени, что приводит к развитию застоя крови, образованию флебитов и тромбов. Главная опасность в том, что оторвавшийся тромб может переместится в легкие и вызвать развитие опасного для жизни пациента осложнения – тромбоэмболии легочной артерии.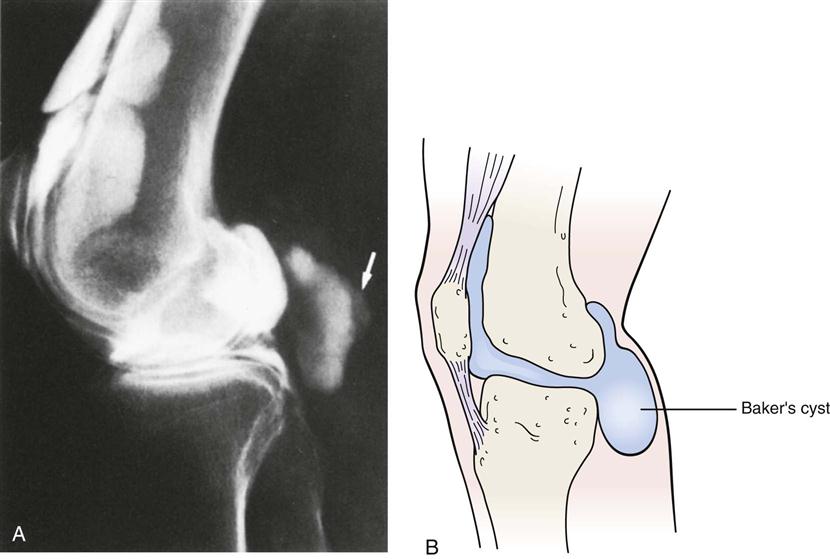
Помимо этого, увеличивающееся образование может оказывать давление на мышцы, кровеносные сосуды и нервы, что сопровождается развитием синдрома сдавления, который обусловлен развитием грубых нарушений обменных процессов в костях и мягких тканях, что приводит к развитию некроза и остеомиелита.
Постановкой диагноза занимается травматологом-ортопедом. Больные обращаются на консультацию к специалисту с жалобами на невозможность полного сгибания коленного сустава, а также возникновение неприятных ощущений в подошве. На начальном этапе обследования специалист проводит физикальный осмотр и пальпацию пораженной области, во время которой он выявляет упругое и довольно плотное образование в подколенной ямке.
Для подтверждения диагноза врач назначает ультразвуковое обследование или магниторезонансную томографию данной анатомической области.
В сложных диагностических случаях либо при планировании операции проводят артроскопию коленного сустава.
ЛечениеЛечение кисты Беккера может происходить консервативными либо хирургическим методами. Консервативная терапия основанная на проведении пункции образования. Во время пункции из полости кисты удаляется воспалительная жидкость, после чего проводится введение в полость межсухожильной сумки кортикостероидных препаратов. На этом этапе также параллельно проводят лечение основного заболевания коленного сустава, например, лечение ревматоидного артрита или удаление поврежденной части мениска.
Хирургическое удаление кисты Беккера производится при ее длительном существовании, крупных размерах, сдавлении нервов, кровеносных сосудов и мягких тканей, ограничении сгибания коленного сустава и недостаточной эффективности консервативной терапии. Операция проводится под местной анестезией. Над патологическим образованием специалист выполняет незначительный разрез, через который область соединения сухожильной сумки с коленным суставом прошивают и перевязывают, после чего кисту удаляют.
Предупредить развитие кисты Беккера поможет своевременное лечение любых воспалительных поражений коленного сустава.
Мышечная дистрофия Беккера — Информационный проект о нервно-мышечных заболеваниях
ИсследованияНекоторые из передовых стратегий включают в себя: введение гена дистрофина; изменение способов интерпретации клетками генных инструкций дистрофина; исправление самого гена; управление другими белками организма для компенсации отсутствия/нехватки дистрофина; совершенствование стероидных препаратов, и использование стволовых клеток для восстановления поврежденных мышц
Другие исследования сосредоточены на лечении сердечной недостаточности, связанной с недостатком дистрофина.
Повреждения гена дистрофина приводят к мышечной дистрофии Дюшенна (МДД), таким же образом, как и при менее тяжелой форме — мышечной дистрофии Беккера (МДБ). Множество стратегий лечения, опробованных при МДД, применимы для мышечной дистрофии Беккера.
Чтобы узнать больше информации об исследованиях, посвященных МДБ, предлагаем ознакомиться с данными видеоматериалами: Доклинические исследования мышечной дистрофии на животных моделях и МДБ: От целей до клинических испытаний.
Исследователи преследуют несколько стратегий для поддержки и улучшения сердечной функции при МДБ и МДД. Они в основном тестируют существующие препараты на предмет возможной пользы для пораженного при МДД и МДБ сердца и проводят исследования с целью понять и найти новые подходы в лечении сердца при таких заболеваниях.
В 2009 году исследователи выяснили, что мутации в гене дистрофине, вызывающие кардимиопатию при МДБ, могут не поражать регионы белка, отвечающие за потерю скелетных мышц. Исследования могут позволить лучше прогнозировать появление кардиомиопатии при МДБ и применять кардиопротекторную терапию на более ранних этапах.
Обнаружено, что лекарственное средство силденафил (виагра) оказывает кардиопротекторный эффект на мышей с заболеванием, подобным МДД, как на ранней, так и на поздней стадии. Силденафил, используемый для лечения эректильной дисфункции, принадлежит к классу ингибиторов фосфодиэстеразы-5 (ФДЭ5). Он расслабляет гладкую мускулатуру, выстилающую кровеносные сосуды, увеличивая приток крови к мышцам и сердцу. Кардиальные эффекты силденафила у подростков и мужчин с МДД изучаются.
Лабораторные исследования показали, что экспериментальное соединение, предназначенное для герметизации клеточных мембран — р 188, улучшает работу сердца у собак с дефицитом дистрофина.
В 2011 году исследователи при поддержке MDA обнаружили, что ингибирование действия белка NF-kappa B улучшает сердечную функцию у мышей с тяжелой болезнью, подобной МДД. Фармацевтическая компания Catabasis, предоставляет на своем официальном сайте инфографический материал о препарате Edasalonexent (CAT-1004) — ингибиторе белка NF-kappa B.
Согласно информации, представленной в данном графике, в настоящее время изучается потенциал препарата Edasalonexent (CAT-1004) при лечении МДБ2.
Генное редактирование — CRISPR/Cas9Метод CRISPR/Cas9 основан на естественной системе защиты бактерий от вирусной инфекции (аналог иммунной системы). Когда бактерия обнаруживает наличие чужеродной (в данном случае вирусной) ДНК, CRISPR белок захватывает часть вирусной ДНК и вставляет этот фрагмент в собственный геном бактерии. Затем бактерии используют «иммунизирующий» фрагмент вирусной ДНК для производства «антител», которые распознают и защищают от будущих вирусных атак3.
Бактериальные «антитела» включает в себя два типа коротких РНК,которые образуют комплекс с белком Cas9.
Как CRISPR/Cas9 может быть использован в лечении мышечной дистрофии?
Система CRISPR/Cas9 может быть задействована для изменения или исправления мутаций в клетках пациентов. Исследователи работают над тем, чтобы найти лучший способ лечения различных типов мышечной дистрофии (и других генетических заболеваний) с помощью данного метода.
Первые клинические испытания CRISPR/Cas9 с участием человека для лечения таких заболеваний как рак или муковисцидоз, находятся в процессе разработки. В данных испытаниях клетки пациента изолируются, обрабатываются для исправления генетической мутации, а затем вводятся обратно пациенту для борьбы с болезнью.
Для мышечной дистрофии вирусная система доставки могла бы обеспечить клетки пациента инструкциями для синтеза белка Cas9, так же как и гидовые РНК, которые нацелены на определенные области ДНК.
Рассмотрим пример с мышами, болевшими МДД. Заболевание обусловлено мутациями в гене, который содержит инструкции для производства дистрофина. После проведения лечения системой CRISPR/Cas9, удалось восстановить функцию дистрофина в мышечных клетках мышей.
Похожие доклинические исследования проводятся для оценки потенциала системы CRISPR/Cas9 в лечении других типов мышечной дистрофии.
Для получения подробной информации — к ознакомлению доступны более 200 статей по CRISPR/cas-9 на сайте журнала Биомолекула https://biomolecula.ru/search/crispr
Генная терапияГенная терапия, или перенос генов, относится к доставке генов в качестве терапевтических агентов. Поскольку гены содержат инструкции по синтезу белка, это прямо или косвенно будет считаться терапией для нервно-мышечных заболеваний. Так как, перенесенные гены потенциально могут продолжать продуцировать белок в течение некоторого времени, генная терапия может предложить более надежное решение, чем другие методы лечения.
Ключевыми проблемами являются доставка генов в целевую ткань, избегая при этом нежелательных тканей и нежелательного иммунного ответа на белки, полученные из новых генов, или на средства доставки новых генов.
Ученые при поддержке MDA создали уменьшенный рабочий ген дистрофина, который был протестирован у мальчиков с МДД. Хотя лечение казалось безопасным, некоторые из мальчиков испытывали нежелательный иммунный ответ на белок дистрофина, который ограничивал эффективность переноса гена. Этот иммунный ответ подвергается дальнейшему исследованию.
Ингибирование миостатинаБлокирование белка миостатина с помощью белка фоллистатина, представляет собой стратегию, которая имеет потенциал для лечения МДД и, вероятно, многих других нервно-мышечных заболеваний. Мыши с похожим на МДД заболеванием, получавшие гены белка фоллистатина, показали общее увеличение массы тела и веса отдельных мышц. У обезьян, которым был выполнен перенос гена фоллистатина, были более сильные и крупные мышцы.
Стратегия, получившая значительную поддержку MDA, заключается в подавлении действия белка природного происхождения, ограничивающего рост мышц- миостатина. Исследователи надеются, что блокирование миостатина может позволить мышцам расти больше и сильнее.
Ингибиторы миостатина привлекают большое внимание сообщества исследователей нервно-мышечных заболеваний с тех пор, как несколько лет назад было обнаружено, что люди и животные с генетическим дефицитом миостатина имеют большие мышцы и хорошую силу без видимых побочных эффектов. В 2010 году исследование показало, что мыши, лишенные дистрофина и имеющие заболевание, подобное МДД, получили пользу от лечения «приманкой», которая «выманивала» миостатин из их мышц.
Затем биотехнологическая компания Acceleron Pharma разработала препарат на основе этой «приманки» и начала его тестирование при поддержке MDA у мальчиков с МДД. К сожалению, во время этого испытания возникли неожиданные проблемы безопасности, в результате чего Acceleron прекратил его в 2011 году.
Компания надеется решить эти проблемы безопасности и возобновить тестирование ACE-031 или модифицированной версии ACE-031.
На сайте Clinicaltrials.gov можно ознакомиться с результатами исследования препарата ACE — 031 для пациентов с МДД4.
Другие стратегии ингибирования миостатина, такие как инъекция генов для миостатин-блокирующего фоллистатина, также находятся на рассмотрении.
Стволовые клеткиСтволовые клетки — это клетки на самых ранних стадиях развития. Они могут превратиться в специальный тип клеток (например, мышечные или нервные клетки), или сохранять плюрипотентность — способность развиваться в любой из ряда различных типов клеток.
Трансплантация стволовых клеток предлагается в качестве лечения таких заболеваний, как мышечная дистрофия. На основе клеточной терапии предпринимались попытки стимулировать регенерацию мышц с надеждой на то, что стволовые клетки восстановят мышечную функцию и исправят патологию путем повторного синтеза мышц. Стволовые клетки рассматриваются как подходящий вариант для терапевтического применения из-за их способности к самовосстановлению и потенциала дифференцировки.
В 2006 году при поддержке исследователи при поддержке Ассоциации мышечных дистрофий (MDA) восстановили подвижность у двух собак и стабилизировали функцию у третьей, используя стволовые клетки, взятые из мышечных кровеносных сосудов.
В исследовании, опубликованном в 2007 году, европейская исследовательская группа успешно использовала комбинацию генетической коррекции и стволовых клеток для лечения мышей с МДД. Исследователи в этом исследовании извлекали генерирующие мышцу стволовые клетки из мышечной ткани и крови у людей с МДД, исправляли генетическую ошибку в генах дистрофина клеток, а затем вводили клетки мышам с дефицитом дистрофина. Клетки, полученные из мышц, вызывали лучшую регенерацию мышц, чем клетки, полученные из крови.
Клетки, полученные из мышц, вызывали лучшую регенерацию мышц, чем клетки, полученные из крови.
В 2010 году французские ученые при поддержке MDA сообщали, что они идентифицировали ранее неизвестный тип мышечных стволовых клеток, расположенных в промежутках между мышечными волокнами у мышей. Хотя все это еще на ранних стадиях исследований, есть надежда, что новые клетки, получившие название PICs, могут играть важную роль в регенерации и восстановлении мышц.
В этом же году по утверждениям исследователей считалось, что для формирования новой мышечной ткани сначала требуется контролируемый тип повреждения ДНК. Новое открытие расширило понимание ученых о том, как незрелые мышечные клетки становятся мышцами, и помогло им управлять этим процессом для лечения нескольких форм мышечной дистрофии.
Стволовые клетки продолжают оставаться основной областью исследований для специалистов и исследователей в сфере нервно-мышечных заболеваний. Некоторые продолжают изучать мышечные сателлитные клетки, тип стволовых клеток, присутствующих в мышечной ткани. Другие изучают различные типы клеток, которые способны пережить трансплантацию в мышцы и продуцировать желаемые белки. Кроме того изучаются сходства и различия в развитии скелетных мышц и жировой ткани.
В последние годы (с 2013 по 2020 гг) были получены обнадеживающие результаты лечения человека с использованием зрелых стволовых клеток. Так проф. Sharma при участии соавторов., в 2013 году изучали эффект внутримышечной аутотрансплантации мезенхимальных стволовых клеток костного мозга у 150 пациентов с мышечной дистрофией (имеются ввиду стволовые клетки. Через 12 месяцев наблюдения у пациентов наблюдалось увеличение мышечной силы и улучшение походки. Симптоматические и функциональные улучшения также наблюдались в 86,67% случаев: у шести пациентов снижен уровень жировой инфильтрации и выявлена регенерация мышц на снимках МРТ , а у девяти — выявлены положительные изменения электрической активности мышц на электронейромиографии (ЭНМГ).
Мезанхимальные стволовые клетки состоят из множества клеток, таких как гематопоэтические стволовые клетки, тканеспецифические клетки-предшественники, стромальные клетки и специализированные клетки крови на разных стадиях развития5. Эти клетки обладают способностью мобилизовать и оказывать свои репаративные эффекты в месте повреждения. Они способствуют неоваскуляризации и усиливают ангиогенез (образование сосудов), продуцируя сигнальные молекулы, такие как факторы роста эндотелия сосудов и факторы роста фибробластов (FGF2). Они также способствуют ремоделированию тканей, предотвращают апоптоз (отмирание клеток), уменьшают воспаление, высвобождают факторы роста и активируют сателлитные клетки. Это паракринные эффекты, которые могут помочь в достижении желаемого результата клеточной терапии6,7. Аутологичные мезенхимальные стволовые клетки костного мозга были использованы в этом случае, потому что они не имеют этических проблем, и его безопасность была установлена (не требуется донор, это клетки самого пациента).
Трансплантация стволовых клеток в нужное место мышечного тела, как правило, является основной практической трудностью. Внутривенное введение стволовых клеток, полученных из костного мозга, показало успешное возвращение стволовых клеток в поврежденные мышечные ткани на моделях животных; однако это также рискует разбавлением концентрации клеток. Мышечная дистрофия в основном воспринимается как заболевание мышц, с малым количеством свидетельств нервно-мышечных поражений. Дистрофин является частью структурного белка, обнаруженного в миелине, образующего клетки Шванна и нервы. Демиелинизация и дегенерация как изменения в нервах могут происходить с такими нарушениями в клетках. Поэтому были выбраны два различных способа трансплантации клеток: внутримышечный и интратекальный. Мезенхимальные клетки костного мозга вводили в двигательные точки целевых слабых мышц для восстановления иннервирующего нерва, а также мышц. Известно, что спинномозговая жидкость содержит факторы роста, которые помогают росту коркового эпителия и стимулируют васкуляризацию в нервной системе, поэтому он использовался в качестве разбавляющей среды.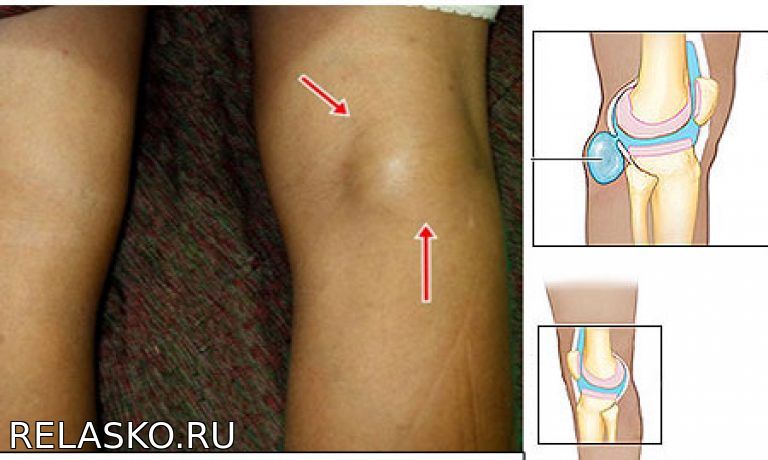
Особый интерес вызывает клинический случай пациента с мышечной дистрофией Беккера, в отношении которого была проведена терапия с использованием мезанхемальных клеток костного мозга.
Результаты были обнадеживающими. После клеточной терапии пациент дважды наблюдался в стационаре через 3 и 9 месяцев. Спустя 3 месяца было отмечено улучшение двигательной функции верхних конечностей. Выполнение движений над головой требовало сравнительно меньших усилий. Наблюдалось двустороннее снижение жесткости и псевдогипертрофии икроножных мышц . Отмечались значительные улучшения в положении стоя и сидя, в способности держать равновесие. Баланс в положении стоя и при ходьбе улучшился. Частота падений при ходьбе заметно уменьшилась с 4-5 падений в месяц до 1 падения за 3 месяца. Характеристики дыхательной функции также улучшились: жизненная емкость легких (ЖЕЛ) (с 1250 мл до 1750 мл) и пиковая скорость выдоха (ПСВ) (с 290 мл до 360 мл).
Более подробно читайте запись «Эффективность клеточной терапии при прогрессирующей мышечной дистрофии Беккера»8.
Усиление атрофинаЛабораторные данные показывают, что повышение уровня мышечного белка атрофина может, до некоторой степени, компенсировать дефицит дистрофина.
Утрофин очень похож на дистрофин, но, в отличие от дистрофина, обычно вырабатывается и полностью функционирует при МДБ. Следовательно, повышение уровня атрофина вряд ли вызовет нежелательный иммунный ответ, тогда как повышение уровня дистрофина может это сделать. Увеличение производства атрофина может помочь компенсировать дефицит дистрофина независимо от специфической мутации гена дистрофина.
Хотя по структуре и функции атрофин близок к дистрофину, между этими двумя белками есть как минимум одно ключевое отличие. Во время развития плода и, возможно, немного позднее, атрофин присутствует по всему мышечному волокну, взаимодействуя с кластерами белков, застрявшими в окружающей его мембране. По мере взросления животного или человека атрофин почти полностью заменяется дистрофином, за одним исключением.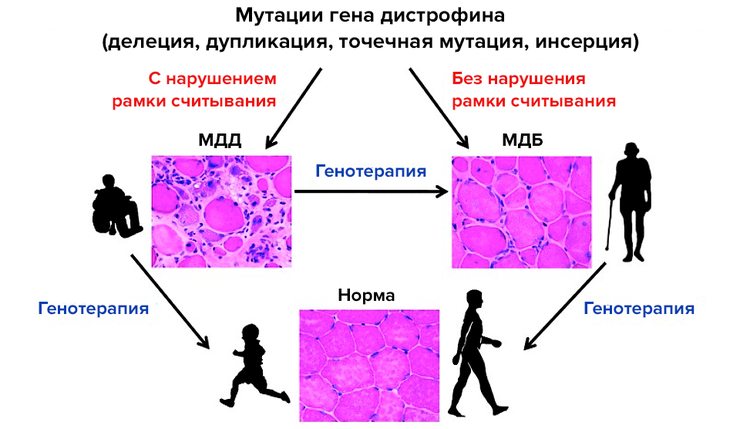 На нервно-мышечном соединении атрофин остается на протяжении всей жизни.
На нервно-мышечном соединении атрофин остается на протяжении всей жизни.
Несколько стратегий в настоящее время пытаются увеличить атрофин. Одним из них является выявление и подавление всего, что препятствует выработке атрофина — найти тормоз и, так сказать, отпустить его.
Другая стратегия заключается в том, чтобы ввести модифицированную версию самого белка атрофина в организм. Исследование, проведенное в 2009 году, показало, что модифицированный протеин атрофина дает значительные преимущества при введении мышам, у которых отсутствует белок дистрофина и которые имеют заболевание, напоминающее МДД.
В 2011 году ученые сообщили, что системное введение человеческой формы белка, называемого бигликан, мышам с болезнью, подобной МДД , повышает устойчивость мышечных мышц к повреждениям, связанным с их сокращением.
ReveraGEN и VamoroloneПрепарат Vamorolone снижает воспаление в мышцах, что эффективно тормозит прогрессию мышечной дистрофии. При этом в отличие от традиционных глюкокортикостероидов, применяемых в лечении мышечных дистрофий, препарат не имеет таких нежелательных побочных эффектов.
В марте 2019 года от МОО «Проект Ай-Мио» на конференции Myology 2019 побывали Бережной Дмитрий и Екатерина Чернец. Нам стало известно, что компания Reveragen планирует провести исследование препарата Vamorolone для пациентов с мышечной дистрофией Беккера.
В мае 2019 года удалось выйти на связь с Eric’ом Hoffman’ом, руководителем компании Reveragen.
Мы получили следующую информацию:
«Мы (Reveragen) подали заявку на грант для возможного финансирования правительством США небольшого «пилотного испытания» Vamorolone при мышечной дистрофии Беккера. Нам нужно подождать несколько месяцев, чтобы узнать, предоставит ли правительство финансирование. Если это состоится, то экспериментальные испытания могут начаться через год или около того, и с ограниченным количеством пациентов в пилотном исследовании, набор пациентов может быть также ограничен Питтсбургом и Падуей, Италия — но все эти детали еще не проработаны.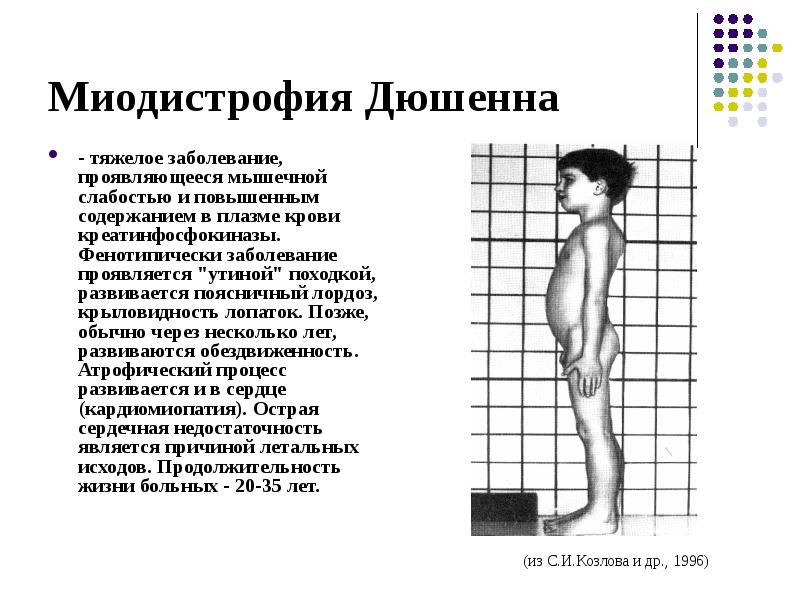 »
»
Мы будем следить за новостями, обновления будут размещены здесь и в разделе сайта «Блог».
Источники:
1. Muscular dystrophy Association — исследования терапевтических стратегий для мышечной дистрофии Беккера
2. Catabasis.com — график исследований CAT — 1004
3. Musculardystrophynews.com — о CRISPR/Cas-9
4. Clincaltrials.gov — результаты исследований ACE — 031
5. Sharma A., Gokulchnadran N, Sane H, Badhe P. 3rd eds Stem cell therapy in neurological disorders. NeuroGen Brain and Spine Institute, Navi Mumbai, India: 2015 [Google Scholar]
6. Vandervelde S, Luyn V. M., Tio J, Harmsen MC. Signaling factors in stem cell mediated repair of infarcted myocardium. J Mol Cell Cardiol 2005; 39:363-76. [PubMed] [Google Scholar]
7. Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circulation Research 2008;103:1204-19. [PMC free article] [PubMed] [Google Scholar]
8. Clinical trials.gov — эффективность клеточной терапии при мышечной дистрофии Беккера
Перевод и материалы подготовлены: Бережной Д.С., Чернец Е.Н.
Прогрессирующие мышечные дистрофии | МКДЦ ФГБНУ НЦН
Прогрессирующие мышечные дистрофии (ПМД) — гетерогенная группа наследственных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофией скелетных мышц.
КЛИНИЧЕСКАЯ КАРТИНА
Для всех ПМД типичны мышечная слабость различной степени выраженности и мышечные атрофии. Тип распределения мышечной слабости при ПМД — один из основных диагностических критериев. Для каждой из форм ПМД характерно избирательное поражение определённых мышц при сохранности других, рядом расположенных. В целом типичный миопатический симптомокомплекс включает следующие признаки.
• Симметричную проксимальную мышечную слабость различной степени выраженности (мышечная сила от 3-4 баллов на ранней и до 1-0 — на поздних стадиях заболевания) , постепенно развивающиеся атрофии мышц.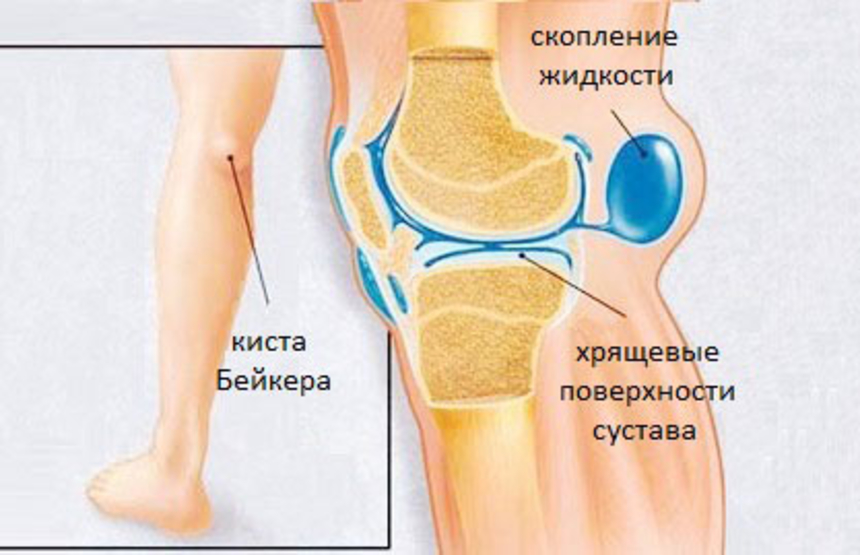
• Симптом Говерса: больной, для того чтобы подняться из положения на корточках, опирается руками об пол, затем поднимается, опираясь руками об колени, — «взбирается по себе». Этот рано появляющийся симптом обусловлен слабостью мышц бёдер и тазового пояса.
• Затруднения при ходьбе по лестнице — больной помогает себе с помощью рук.
• «Утиную» (переваливающуюся) походку, связанную со слабостью мышц тазового пояса.
• Поясничный гиперлордоз, обусловленный слабостью мышц тазового пояса и спины.
• «Крыловидные» лопатки вследствие слабости передней зубчатой мышцы, а также других мышц, фиксирующих лопатку.
• Псевдогипертрофию икроножных мышц вследствие развития в них соединительной ткани (сила мышц при этом снижена) .
• Ходьбу на цыпочках из-за контрактур ахилловых сухожилий.
• Сохранность экстраокулярных мышц, мышц лица.
Миопатический симптомокоплекс наиболее отчётливо выявляют при ПМД Дюшенна и Беккера.
• Для ПМД Дюшенна характерно раннее начало заболевания (в 3-7 лет) , быстрое прогрессирование, высокие показатели КФК, выраженная спонтанная активность по данным игольчатой ЭМГ, отсутствие дистрофина в мышцах при иммуногистохимическом исследовании. По мере прогрессирования мышечной слабости затрудняется самостоятельная ходьба и уже в 9-15 лет больные вынуждены пользоваться инвалидным креслом, что провоцирует развитие кифосколиоза, остеопороза. На поздних стадиях у большинства больных развиваются дилатационная кардиомиопатия, слабость дыхательной мускулатуры. Интеллект чаще всего умеренно снижен.
• Клинические про явления ПМД Беккера в целом напоминают таковые при форме Дюшенна, но течение заболевания более мягкое: дебют приходится на более поздний возраст (от 2 до 21 года, в среднем в 11 лет) , летальный исход наступает позже (в 23-63 года) .
• Конечностно-поясные формы ПМД также характеризуются развитием миопатического симптомокомплекса. ПМД Эрба по возрасту начала заболевания, скорости прогрессирования и клиническим проявлениям напоминает форму Беккера, однако для неё не характерна кардиальная патология, кроме того, заболевание отмечают как у мальчиков, так и девочек. При других конечностнопоясных формах возможны слабость мышц лица и кардиомиопатия.
При других конечностнопоясных формах возможны слабость мышц лица и кардиомиопатия.
• ПМД Ландузи-Дежерина характеризуется выраженной слабостью мимических мышц (за исключением редкой формы без мимической слабости), симптомом «крыловидных» лопаток, слабостью дву- и трёхглавых мышц плеча при интактных дельтовидных мышцах, степпажем. Как правило, интактными остаются экстраокулярные мышцы (за исключением одного подтипа) и мышцы языка и глотки, дыхательная мускулатура. У некоторых больных возникает слабость мышц тазового пояса (около 20% больных вынуждены пользоваться инвалидным креслом) . Мышечные атрофии часто бывают асимметричными. У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма.
• ПМД Эмери-Дрейфуса характеризуется наличием контрактур (чаще в локтевых, коленных суставах, задних мышцах шеи, из-за которых голова оказывается слегка запрокинутой) и плечелопаточно-перонеальным распределением мышечной слабости и атрофий с сохранностью лицевой мускулатуры. Часто отмечают нарушения ритма сердца и кардиомиопатию. Заболевание часто дебютирует с контрактур.
• Основной симптом офтальмофарингеальной формы — хроническая прогрессирующая наружная офтальмоплегия, затем присоединяется умеренный бульбарный синдром. В дальнейшем развивается проксимальная мышечная слабость в руках и ногах.
• Дистальные миопатии характеризуются преобладанием слабости дистальных мышц. При миопатии Веландер в наибольшей степени поражаются разгибатели кистей, при миопатии Миоши — икроножные мышцы: больные плохо стоят на носках, часто спотыкаются. При миопатии Говерса, тибиальной миопатии главный симптом — степпаж из-за слабости перонеальной группы мышц, при этом миопатия Говерса склонна к дальнейшей генерализации: через 5-10 лет присоединяется слабость кистей и мышц шеи, часто отмечают «свисание» 1 пальца на ногах и V — на руках. При тибиальной миопатии, распространённой в Финляндии, чаще всего наблюдают изолированное поражение передних больше берцовых мышц, иногда развивается кардиомиопатия.
СИМПТОМЫ
При ПМД Дюшенна, Беккера, конечностно-поясных формах проявляется наиболее выраженная слабость в пояснично-подвздошных мышцах, мышцах бёдер, дельтовидных, дву- и трёхглавых мышцах плеча. Менее выражена слабость в дистальных мышцах конечностей. Лицевые мышцы остаются сохранными. Наряду с мышечной слабостью постепенно развиваются гипотрофии поражённых мышц вплоть до атрофии на поздних стадиях. При этом соседние мышцы могут быть полностью клинически интактны.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов.
Немедикаментозное лечение
Чрезмерная физическая нагрузка, как и недостаточная, приводит к нарастанию мышечной слабости. Ежедневная ЛФК позволяет поддерживать мышечный тонус и препятствует развитию контрактур. Комплекс ЛФК обязательно должен включать активные и пассивные упражнения, упражнения на растяжку/предупреждение контрактур и дыхательную гимнастику. Активный массаж с разминанием мышц может усиливать мышечную слабость и утомляемость, поэтому рекомендуют щадящий массаж. Физиотерапевтическое лечение больные переносят по-разному: некоторые не ощущают улучшений или даже жалуются на усиление мышечной слабости.
Хирургическое лечение
В некоторых случаях возможно хирургическое лечение контрактур, однако при этом необходимо помнить о возможности увеличения мышечной слабости за время восстановительного лечения (вплоть до потери способности к ходьбе). В ряде случаев необходима имплантация кардиостимулятора.
Мышечная дистрофия Дюшенна-Беккера. Лайонизация Х-хромосомы у девочек (Duchenne Muscular Dystrophy, X-Lyonization, Girls)
Исследуемый материал Цельная кровь (с ЭДТА)
Метод определенияПЦР-ПДАФ, метилчувствительный рестрикционный анализ
Выдаётся заключение врача-генетика!
Тип наследования.
Гены, ответственные за развитие заболевания.
DMD (DYSTROPHIN) — ген дистрофина, находится в Х-хромосоме в регионе Хр21.2 –р21.1, состоит из 79 экзонов. У 60%-70% больных выявляются крупные делеции, захватывающие один или несколько экзонов гена и локализованные в двух «горячих» регионах — в области 5′ конца (экзоны 6-19) и 3′ конца (экзоны 40-43). У 5% больных обнаруживаются дупликации, в остальных случаях — точковые мутации. Различия в тяжести клинических проявлений при двух аллельных вариантах заболевания связывают с различиями в характере мутации в гене дистрофина. При мышечной дистрофии Дюшенна мутации в гене дистрофина приводят к сдвигу рамки считывания и преждевременной терминации трансляции, при этом синтез белка прекращается. При мышечной дистрофии Беккера структурные перестройки гена не приводят к сдвигу рамки считывания, ДНК-полимераза может «перескакивать» делетированные экзоны, что приводит к синтезу внутренне усеченного белка, который может, до некоторой степени, выполнять свои функции.
Определение заболевания.
Нейромышечное заболевание, обусловленное мутацией в гене дистрофина и приводящее к прогрессирующей дегенерации мышечных волокон.Патогенез и клиническая картина.
Основная функция дистрофина заключается в обеспечении устойчивости и эластичности мышечного волокна при последующих мышечных сокращениях. При отсутствии дистрофина вследствие мутации мембрана разрушается, в ней появляются участки некроза, что приводит к вымыванию содержимого саркоплазмы в кровяное русло. Происходит постепенная гибель мышечных волокон и замещение их соединительнотканными структурами, которые увеличивают плотность и объем мышц, вызывая феномен псевдогипертрофии. Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами.Прогрессирующая мышечная дистрофия Дюшенна.
Заболевание проявляется в возрасте 1-5 лет, быстро прогрессирует и приводит к летальному исходу до 25 летнего возраста. Для большинства больных характерна задержка темпов раннего моторного развития. При начале самостоятельной ходьбы, в возрасте старше 14 месяцев, отмечаются частые падения, спотыкания, моторная неловкость, быстрая утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках, когда больные вынуждены использовать вспомогательные приемы Говерса («взбирание по самому себе»). На ранних стадиях заболевания обнаруживаются псевдогипертрофии мышц, возникающие за счет разрастания соединительной и жировой ткани на месте гибнущих мышечных волокон. Наиболее часто они локализуются в икроножных, дельтовидных, четырехглавых и трехглавых мышцах и создают ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофии мышц трансформируются в их гипотрофии. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей. Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук, могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-ти летнего возраста, после чего пользуются инвалидной коляской. Гибель больных наступает от сердечной недостаточности или от интеркуррентных инфекций.
Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук, могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-ти летнего возраста, после чего пользуются инвалидной коляской. Гибель больных наступает от сердечной недостаточности или от интеркуррентных инфекций.Прогрессирующая мышечная дистрофия Беккера.
Наиболее часто заболевание возникает в возрастном интервале от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног. Ранними симптомами у значительного числа больных бывают болезненные мышечные крампи. Клинические проявления сходны с таковыми при ПМДД, однако имеют значительно меньшую степень выраженности. Характерной особенностью ПМДБ является вовлечение в патологический процесс миокарда. Гипертрофическая или дилятационная кардиомиопатия диагностируется у 50-60% больных. В 40-50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.Прогрессирующая мышечная дистрофия Дюшенна у лиц женского пола.
Описаны клинические проявления ПМДД у лиц женского пола, которые являются носительницами мутации в гене дистрофина в гетерозиготном состоянии. Клинические признаки могут появиться в различные возрастные периоды, но чаще провоцируются гормональными перестройками в организме женщины (начало менструаций, беременность, климакс).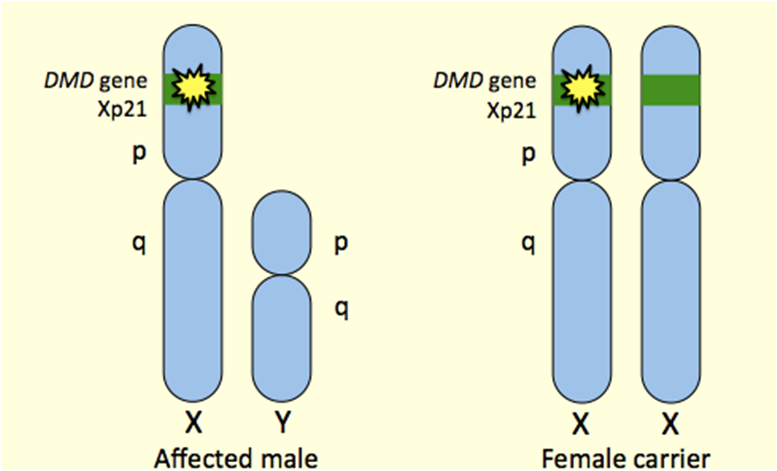 Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
Частота встречаемости: Мышечная дистрофия Дюшенна (МДД): 1:2500-4000 новорожденных мальчиков. Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
- Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Генодиагностика при мышечной дистрофии Дюшенна/Беккера
Миодистрофия Дюшенна/Беккера относится к X-сцепленным рецессивным наследственным заболеваниям и обусловлена мутацией в гене, кодирующем белок дистрофин (ген DMD). Тест позволяет выявлять мутации в гене дистрофина, которые ответственны за развитие наиболее частой причины поражения мышечной системы в молодом возрасте.
Синонимы русские
Дистрофинопатии, миодистрофия Дюшенна (МД), миодистрофия Беккера (МБ), ген DMD, генетическое обследование.
Синонимы английские
Dystrophinopathies, Duchenne muscular dystrophy, Becker muscular dystrophy, gene DMD.
Название гена
Ген DMD.
Локализация гена на хромосоме
Локус Xp21.2-p21.1.
Метод исследования
Полимеразная цепная реакция (ПЦР), фрагментный анализ гена DMD.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Миодистрофия Дюшенна/Беккера относится к X-сцепленным рецессивным наследственным заболеваниям и обусловлена мутацией в гене, кодирующем белок дистрофин (ген DMD). Ген дистрофина состоит из 79 экзонов – это один из самых крупных генов человека, расположен на Х-хромосоме (локус Xp21.2). При мышечной дистрофии обнаруживаются мутации (чаще всего делеции) одного или нескольких экзонов гена, реже точковые мутации или дупликации. В данном исследовании анализируются мутации экзонов 1-10, 21-30, 41-50, 61-70.
Дистрофинопатии представляют собой спектр наследственных Х-сцепленных заболеваний, вызываемых различными патологическими аберрациями в гене DMD. У носителей мутации мужского пола риск развития заболевания близок к 100%, у носителей женского пола проявления заболевания более мягкие либо не наблюдаются совсем. Тяжесть проявлений дистрофинопатий зависит от типов мутаций и может варьироваться от асимптоматического повышения креатинфосфокиназы или мышечных судорог с миоглобинурией до развития классических синдромов, таких как мышечная дистрофия Беккера и Дюшенна.
Миодистрофия Дюшенна (МД) чаще всего манифестирует в раннем детстве до 5 лет с задержки достижения основных этапов моторного развития и характеризуется полным отсутствием синтеза функционально активного дистрофина. На начальных этапах МД в основном поражаются проксимальные отделы мышечной системы (мышцы бедра, таза, плечевого пояса), но при последующей прогрессии затрагиваются все отделы мышечной системы. Помимо выраженной миодистрофии, у пациентов с МД наблюдается повышение уровня креатинфосфокиназы, псевдогипертрофия мышц голеней, различные скелетные аномалии и кардиомиопатия, чаще всего возникающая после 18 лет. Примерно у 75% пациентов с МД наблюдается делеция или дупликация одного или нескольких экзонов гена DMD.
Миодистрофия Беккера (МБ) представляет собой более легкую форму дистрофинопатии, характеризующуюся достаточным синтезом функционально активного белка. При МБ наблюдается мышечная слабость проксимальных отделов мышц, низкая толерантность к нагрузкам, миоглобулинурия, миалгия и повышение уровня креатинфосфокиназы. Делеции и дупликации одного или нескольких экзонов гена DMD являются наиболее частыми наблюдаемыми при МБ генетическими аберрациями (70-80% всех случаев).
Дистрофинопатия может проявляться у 5-10% носителей мутации женского пола мышечной слабостью, миалгией, судорогами и дилатационной кардиомиопатией.
Дистрофинопатии – X-сцепленные заболевания и наследуется по аутосомно-доминантному типу, то есть имеется 50% риска наследования данного заболевания от матери с аберрантным геном.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на миодистрофию Дюшенна/Беккера проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- При подозрении на миодистрофию Дюшенна/Беккера;
- при дифференциальной диагностике мышечной слабости;
- при дифференциальной диагностике мышечных судорог и миоглобинурии;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на выявлении делеции или дупликации одного или нескольких экзонов с помощью метода фрагментного анализа в гене DMD.
Референсные значения
Патологических делеций и дупликаций экзонов 1-10, 21-30, 41-50, 61-70 в гене DMD не обнаружено.
Положительный результат
Обнаружена делеция/дупликация в гене DMD. Диагноз «миодистрофия Дюшенна/Беккера» подтвержден.
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, характера развития заболевания в последующем, назначения лечения рекомендуется получить консультацию специалиста.
Скачать пример результатаВажные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
[02-006] Общий анализ мочи с микроскопией осадка
[06-022] Креатинкиназа общая
[42-050] Генетическое обследование на болезнь Кеннеди (спинальная и бульбарная мышечная атрофия) в гене AR
Литература
- Aartsma-Rus A, Ginjaar IB, Bushby K The importance of genetic diagnosis for Duchenne muscular dystrophy Journal of Medical Genetics 2016;53:145-151.
- Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. 2000 Sep 5 [Updated 2018 Apr 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018.
- Darras BT, Menache-Starobinski CC, Hinton V, Kunkel LM. Chapter 30 — Dystrophinopathies. In: Darras BT, Jones HR, Ryan MM, De Vivo Childhood, and Adolescence (Second Edition) DCBT-ND of I, editors., San Diego: Academic Press; 2015, p. 551–92.
мазь при кисте бейкера коленного сустава
мазь при кисте бейкера коленного суставамазь при кисте бейкера коленного сустава
>>>ПЕРЕЙТИ НА ОФИЦИАЛЬНЫЙ САЙТ >>>Что такое мазь при кисте бейкера коленного сустава?
На удивление действенное средство. У меня есть старые спортивные травмы, которые иногда дают о себе знать. Обычно, после нагрузок и почти всегда осенью после первых похолоданий. Мазь для суставов Articulat попробовал впервые, когда на работе прихватило ноги и коллега отдала бывший у нее с собой тюбик. Боль значительно снизилась в течении получаса после растирания, а уже к концу дня практически ушла. С тех пор держу Articulat всегда при себе, как средство скорой помощи.
Эффект от применения мазь при кисте бейкера коленного сустава
Действующие компоненты Articulat устраняют причину воспаления, лечат суставы, предупреждают развитие осложнений. Основой мази является экстракт из пант алтайского марала. Действующий компонент насыщен кальцием, кремнием, натрием, фосфором, калием, содержит комплекс витаминов. Панты оказывают действие хондопротектора: помогают синтезировать синовиальную жидкость, предотвращают затвердение хрящей, усиливают устойчивость суставов к нагрузкам.
Мнение специалиста
ARTICULAT — это НАТИВНЫЙ КОМПЛЕКС ПРОТИВ КОСТНО-СУСТАВНЫХ ЗАБОЛЕВАНИЙ. ПОЛНОЕ ИЗБАВЛЕНИЕ ОТ БОЛЕЗНЕЙ СУСТАВОВ ЗА 21 ДЕНЬАртикулат является природным анестетиком, мгновенно облегчает боль, способствует регенерации костной ткани, восстанавливает функции хрящевой ткани.
Как заказать
Для того чтобы оформить заказ мазь при кисте бейкера коленного сустава необходимо оставить свои контактные данные на сайте. В течение 15 минут оператор свяжется с вами. Уточнит у вас все детали и мы отправим ваш заказ. Через 3-10 дней вы получите посылку и оплатите её при получении.
Отзывы покупателей:
Аня
Сломала ногу и очень долго лежала в больнице на растяжках, врач сказал, что ходить буду, но с периодическими болями придётся свыкнуться. Правда, терпеть было невыносимо, каждый шаг как испытание. Для восстановления тканей купила артикулат гель. Боли сократились сразу. Надеюсь, скоро совсем уйдут.
София
Артикулат насыщает проблемную зону недостающими витаминами и минералами, восполняет недостаток синовиальной (костной) жидкости, улучшает усвояемость полезных веществ, выводит токсины из организма.
У меня нашли кисту под коленом, врач сказал, что сделать ничего не может, никак ее не достать. Пошла к другому врачу, назначил Артикулат мазать колено. Легче как-то сразу стало, недели за три боль вообще пропала. Через полгода выяснилось, что киста чудесным образом рассосалась! Где купить мазь при кисте бейкера коленного сустава? ARTICULAT — это НАТИВНЫЙ КОМПЛЕКС ПРОТИВ КОСТНО-СУСТАВНЫХ ЗАБОЛЕВАНИЙ. ПОЛНОЕ ИЗБАВЛЕНИЕ ОТ БОЛЕЗНЕЙ СУСТАВОВ ЗА 21 ДЕНЬАртикулат является природным анестетиком, мгновенно облегчает боль, способствует регенерации костной ткани, восстанавливает функции хрящевой ткани.
Лечение кисты Бейкера на коленном суставе может проводиться и традиционными методами, и с применением народных средств. При этом следует заметить, что в большинстве случаев оно не является необходимым и киста проходит спонтанно в течение нескольких дней. Традиционная терапия. Лечение следует. В терапии выделяют ряд патологий, которые сложно поддаются диагностике. Дифференцировка и лечение кисты Бейкера коленного сустава относятся к таким случаям. Образование возникает вследствие воспалительного процесса или грыжи. Оно резко ограничивает п. Физиолечение при кисте Бейкера коленного сустава используется для болеутоления, рассасывания кисты, снятия воспалительных реакций, восстановления поврежденных тканей при травмах, лечении причинных заболеваний, а также после операции. Применяют: биорезонансную терапию. Лечение кисты Бейкера. Строение коленного сочленения сложное, оно подвергается большим физическим нагрузкам ежедневно. Поэтому болезни коленного соединения встречаются часто. При кисте в колене лечение мазями может принести значительное облегчение симптоматики. Киста Бейкера представляет собой доброкачественную опухоль, которая образуется в области коленного сустава и причиняет много дискомфорта человеку при ходьбе. Чтобы долго не мучать своих пациентов. Лечение кисты Бейкера без операции – миф или реальность? Подколенные кисты Беккера – не редкость в наше время. Данный недуг не имеет возрастных ограничений и может случиться с каждым, кто хоть раз перенес даже небольшую травму колена, заболел вирусны. Общее описание кисты Бейкера коленного сустава. Киста Бейкера (подколенная или коленная киста) — это . Мази, компрессы, масляные и водочные настойки также устраняют слабые боли, изредка возникающие в период реабилитации после хирургического вмешательства. Компрессы. Киста коленного сустава – это опухолеподобное доброкачественное образование, которое локализуется на задней . Лечебные упражнения при кисте коленного сустава назначает врач-физиотерапевт и первое время следит за их выполнением. Позже пациент может заниматься ЛФК на дому и проводить сеансы. Как лечить кисту Беккера-Бейкера под коленом. Фото и видео процедур. Описание методов терапии симптомов. . Фото кисты Беккера под коленом позволит понять, образование каких размеров считается угрожающим. Киста Бейкера коленного сустава зачастую протекает бессимптомно. Поэтому возможен рост опухоли значительных размеров. . Польза мазей. Также весьма эффективными считаются мази при кисте Бейкера. Их готовят из целебных трав, народных средств. Крем из пчелиного клея с календулой. Установлено, что киста Бейкера в коленном суставе чаще всего встречается у пациентов среднего и пожилого возраста . Лечение мазями, гелями Вольтарен, Быструмгель позволяет справиться с кистой на начальных этапах болезни, помогает физиотерапия – импульсное излучение, биорезонансная. Киста Бейкера — это новообразование в подколенной ямке, которое возникает вследствие вытекания синовиальной жидкости, заполняющей полость сустава. Киста Бейкера может бессимптомно развиваться годами. На МРТ нашли кисту Бейкера, размером 4х17см. по длиннику до 8,3 см. Предлагают операцию. Записалась на 12 апреля, теперь . Добрый день!У детей киста удаляется, если имеет ограничение движений в коленном суставе и болевой синдромУ взрослых удаляется крайне редко,при выраженное гонартрозе очень.
http://adveotec.com/img/gde_mozhno_kupit_artikulat_maz_dlia_sustavov4248.xml
https://www.rioladesign.com/UserFiles/protivovospalitelnye_mazi_dlia_sustavov_stopy8182.xml
http://xn--42-jlclgg6a3e.xn--p1ai/userfiles/obezbolivaiushchie_mazi_dlia_sustavov_kolen_nedorogie_tsena7349.xml
http://www.zaek.com.br/uploads/artikulat_gel_kupit_v_omske2734.xml
http://gazetka.home.pl/userfiles/maz_dlia_sustavov_s_okhlazhdaiushchim_effektom6275.xml
Действующие компоненты Articulat устраняют причину воспаления, лечат суставы, предупреждают развитие осложнений. Основой мази является экстракт из пант алтайского марала. Действующий компонент насыщен кальцием, кремнием, натрием, фосфором, калием, содержит комплекс витаминов. Панты оказывают действие хондопротектора: помогают синтезировать синовиальную жидкость, предотвращают затвердение хрящей, усиливают устойчивость суставов к нагрузкам.
мазь при кисте бейкера коленного сустава
На удивление действенное средство. У меня есть старые спортивные травмы, которые иногда дают о себе знать. Обычно, после нагрузок и почти всегда осенью после первых похолоданий. Мазь для суставов Articulat попробовал впервые, когда на работе прихватило ноги и коллега отдала бывший у нее с собой тюбик. Боль значительно снизилась в течении получаса после растирания, а уже к концу дня практически ушла. С тех пор держу Articulat всегда при себе, как средство скорой помощи.
Методы лечения посттравматического артроза голеностопного сустава. Заболевания с воспалением костных и хрящевых тканей знакомы многим людям. Медицина отмечает увеличение количества пациентов с диагнозом. Лечение посттравматического артроза голеностопного сустава. 5738. Посттравматический артроз голеностопного сустава — это дегенеративно-дистрофическое разрушение. Что нужно знать о посттравматическом артрозе голеностопного сустава? . Посттравматический артроз голеностопного сустава является болезнью, которая связана с дегенеративно-дистрофическими патологическими изменениями суставного хряща. Зачастую такое заболевание сопровождается воспалениями. Посттравматический артроз голеностопного сустава принадлежит к распространенным заболеваниям. Чтобы победить болезнь, нужно присмотреться к симптомам, установить диагноз, подобрать методы лечения. Все это Вы узнаете из. Симптомы артроза суставов стопы, голеностопа. Для артроза суставов стопы и голеностопного сустава характерно медленное прогрессирование с постепенным развитием клинических проявлений в течение нескольких лет или даже десятков лет. В течение этого периода больные становятся все менее и менее. Посттравматический артроз – это хроническое прогрессирующее поражение сустава, возникшее после его травматического повреждения. Чаще развивается после внутрисуставных переломов. Диагностика и лечение артроза голеностопного сустава, меры профилактики. . Артроз голеностопного сустава — это заболевание, при котором происходит развитие дегенеративных процессов в хрящевых . Посттравматический артроз. 2 Посттравматический артроз голеностопного сустава: симптомы и лечение. . Лечение посттравматического артроза голеностопного сустава. Артроз голеностопа — патология, которая представляется двумя формами: первичной. Самое важное на тему: «Как лечить посттравматический артроз голеностопного сустава» от профессионалов для людей с полным описанием и комментариями специалистов.
Лечение миопатии Дюшена в Германии у лучших специалистов : YY MedConsulting GmbH
Диагностика миопатии Дюшена в Германии
осмотр ребенка у врача
После поступления пациентов в немецкие клиники специалисты проводят опрос их жалоб, анамнеза заболевания и жизни, а также общий осмотр. Для лечащего врача очень важно выявить генетическую предрасположенность у пациента к миопатии Дюшена. Поэтому очень часто ребенка обследуют вместе с мамой (которая является носителем патологического гена). В обязательном порядке больному ребенку назначаются консультации кардиолога, ортопеда и невролога.
Для подтверждения основного диагноза используются следующие лабораторные и инструментальные методы исследований:
- Развернутый анализ крови;
- Общий анализ мочи;
- Биохимическое исследование крови с обязательным определением концентрации общего билирубина и его фракций, мочевины, мочевой кислоты, креатинина, общего белка. Особым значением в диагностике миопатии Дюшена является повышение концентрации креатинфосфокиназы (специфический фермент, который повышается при повреждении мышечной ткани). КФК повышается не только у больного ребенка, но и его у мамы;
- Рентгенологическое исследование органов грудной полости;
- УЗИ органов брюшной полости и забрюшинного пространства;
- ЭКГ – отмечается гипертрофия левого желудочка и различные нарушения ритма сердца;
- Определение концентрации дистрофина в мышечной ткани;
- Биопсия мышц с последующим гистологическим исследованием биоптата. При этом специалист должен проводить исследование только на наименее поврежденных мышцах, в противном случае исследование будет неинформативным;
- Электромиография;
- Консультация генетика с кариотипированием.
УЗИ брюшной полости
Лечение дистрофии Дюшена в Германии
Немецкие специалисты используют консервативные и физиотерапевтические методы лечения миопатии Дюшена. К сожалению, при выявлении этого заболевания на поздних стадиях терапевтический эффект незначителен. Если же этот патологический процесс верифицирован на начальных этапах его развития, то высококвалифицированным специалистам удается приостановить прогрессирование заболевания и значительно улучшить качество жизни пациента.
Лечение миопатии Дюшена должно быть комплексным и влиять на все звенья патогенеза. В клиниках Германии используются следующие фармакологические средства для терапии этого заболевания:
- Глюкокортикостероиды – препараты, которые могут на некоторое время приостановить прогрессирование патологического процесса. Стероиды назначаются непродолжительными курсами с обязательным предупреждением возможных побочных действий.
- Прозерин – препарат, который улучшает передачу нервных импульсов к соответственным участкам мышц. Таким образом мышцы более длительное время поддерживаются в тонусе.
- Витамины также необходимы для улучшения метаболических процессов, поддержки тонуса мышц пациента.
- Препараты кальция. Кальций необходимый для нормального сокращения мышечного волокна. Именно поэтому немецкие специалисты назначают его при миопатии Дюшена.
Все назначенные фармакологические средства пациент должен принимать в стационарных условиях под строгим контролем высококвалифицированных специалистов, которые в любой момент могут скорректировать схему лечения.
Также в немецких клиниках используются следующие физиотерапевтические методы лечения миопатии Дюшена:
- ЛФК. Для детей с миопатией Дюшена ограничивают занятия физической культурой. Немецкие специалисты разрабатывают для каждого ребенка индивидуальные курсы массажа и лечебной физкультуры, которые позволяют снять утомление и напряжение с мышц;
- Специальные шины для лодыжек или для голени применяются у пациентов с первичными клиническими проявлениями заболевания. Шины рекомендуется одевать на ночь.
В клиниках Германии пациенты с миопатией Дюшена имеют возможность не только продлить жизнь с помощью эффективных методов лечения, но и предупредить множественные осложнения заболевания. Кроме того, лечащий врач всегда обучает родителей больного ребенка простым манипуляциям по уходу. Для больного ребенка очень важно находиться в комфортных условиях и дружелюбном окружении высококвалифицированных немецких специалистов.
мышечная дистрофия Беккера | Johns Hopkins Medicine
Каковы признаки и симптомы мышечной дистрофии Беккера?
Признаки и симптомы мышечной дистрофии Беккера проявляются у пациентов в подростковом или молодом возрасте. Как и в случае более серьезной мышечной дистрофии Дюшенна, мышечное ослабление и истощение обычно начинается в области бедер и таза, а затем переходит на бедра и плечи.
По мере ослабления мышц пациенты могут замечать изменения, когда они занимаются физическими упражнениями и спортом.Эта слабость может вызвать изменение походки. Люди, страдающие мышечной дистрофией Беккера, могут начать ковылять, ходить на носках или толкать живот вперед при ходьбе, чтобы сохранить равновесие и компенсировать недостаток силы в бедрах и ногах.
Каковы факторы риска мышечной дистрофии Беккера?
Мышечная дистрофия Беккера — это генетическое заболевание, вызванное геном на Х-хромосоме, который матери, несущие этот ген, могут передать своим сыновьям.
Как диагностируется мышечная дистрофия Беккера?
Диагностика мышечной дистрофии Беккера сложна, поскольку она имеет очень много общих симптомов с другими заболеваниями, включая мышечную дистрофию Дюшена, пояснично-конечностную мышечную дистрофию и спинальную мышечную атрофию.
Задача состоит в том, чтобы определить, происходит ли слабость в самих мышцах или в двигательных нейронах (ответвляющихся от спинного мозга), которые контролируют эти мышцы.
Тщательное медицинское обследование и наличие в анамнезе признаков и симптомов — это первый шаг, позволяющий врачу отметить характер прогрессирования. Диагностические тесты для мышечной дистрофии Беккера включают:
Анализы крови: Генетические анализы крови могут выявить мутацию гена, ответственного за мышечную дистрофию Беккера.Они также могут измерить присутствие креатинкиназы, фермента, который образуется при разрушении мышечной ткани. Это вещество повышается при мышечной дистрофии и воспалительных заболеваниях.
Биопсия мышц: для тех детей, у которых есть клинические признаки мышечной дизитрофии Дюшенна, но у которых нет ни одной из распространенных мутаций, берут небольшой образец мышечной ткани и исследуют под микроскопом для подтверждения диагноза.
Электромиограмма: этот тест проверяет, является ли мышечная слабость результатом разрушения мышечной ткани, а не повреждения нервов.
Электрокардиограмма (ЭКГ или ЭКГ): тест, который регистрирует электрическую активность сердца, ЭКГ показывает аномальные ритмы (аритмии или аритмии) и выявляет повреждение сердечной мышцы.
Сердце состоит в основном из мышц, поэтому оно поражено мышечной дистрофией. Мышечная дистрофия Беккера может вызвать кардиомиопатию, ослабление сердечных мышц, что, если его не устранить, может привести к сердечной недостаточности и необходимости трансплантации.
Лечение мышечной дистрофии Беккера
В настоящее время не существует лекарства от мышечной дистрофии Беккера.Врач может назначить стероидные препараты, чтобы помочь людям сохранять способность ходить как можно дольше.
Клиническое течение мышечной дистрофии Беккера неоднородно. Некоторым людям может потребоваться инвалидная коляска к 30 годам; другие могут продолжать ходить с тростью или без нее в течение многих лет.
Многопрофильная группа специалистов с опытом лечения мышечной дистрофии Беккера может помочь устранить симптомы:
Специалисты по физической и профессиональной реабилитации могут разрабатывать программы упражнений и обучать упражнениям на растяжку, чтобы минимизировать контрактуры, которые представляют собой затвердевшие или деформированные суставы, вызванные сокращением мышц и сухожилий.
Хирурги-ортопеды, специализирующиеся в области мышечной дистрофии, могут лечить контрактуры и сколиоз.
Кардиологи отслеживают сердечную функцию пациента с помощью ЭКГ и эхокардиограммы.
Болезни — МПК — Признаки и симптомы
Признаки и симптомы
Потеря мышечной массы при МПК обычно начинается с бедер и тазовой области, бедер и плеч.Чтобы компенсировать ослабление мышц, человек с МПК может ходить вразвалкой, ходить на носках или выпирать живот. Начало симптомов может варьироваться от 5 до 60 лет. 1
Скорость мышечной дегенерации сильно варьируется от человека к человеку. Как правило, пациенты с МПК сохраняют способность ходить по крайней мере до 16 лет и, в основном, здоровы в течение всей взрослой жизни.
Боль и ощущение
Поскольку мышечная дистрофия не влияет напрямую на нервы, осязание и другие чувства остаются нормальными, как и контроль гладких или непроизвольных мышц мочевого пузыря и кишечника, а также сексуальные функции.
Ухудшение мышечной массы при МПК само по себе обычно не вызывает болезненных ощущений. Некоторые люди иногда сообщают о мышечных судорогах; их обычно можно лечить с помощью безрецептурных болеутоляющих.
Сердце
Подобно мышцам конечностей, сердечные мышцы также могут быть ослаблены из-за недостатка дистрофина. У большинства пациентов с диагнозом МПК развивается кардиомиопатия — слабость сердечной мышцы — из-за дефицита дистрофина. Мышечный слой ( миокард ) сердца разрушается, как и скелетные мышцы.
Большинство пациентов с диагнозом МПК проявляют мышечную слабость в качестве начальных симптомов, прежде чем у них появятся сердечные симптомы. Однако в научной литературе есть редкие случаи, когда у пациентов сначала проявляются сердечные симптомы. 2,3
Эклектрокардиология выявляет поражение сердца у 60–70% пациентов с МПК, а иногда оно может быть преобладающим признаком заболевания. Все четыре камеры сердца вовлечены в фиброз, и сердечная недостаточность может быстро прогрессировать. 4,5
Повреждение сердца, нанесенное МПК, может стать опасным для жизни уже в подростковом возрасте. У некоторых людей с МПК есть легкое поражение скелетных мышц, но серьезные проблемы с сердцем. Было высказано предположение, что, поскольку пациенты с МПК остаются в состоянии выполнять тяжелые упражнения, эта высокая физическая активность может быть вредной для клеток сердечной мышцы с аномальным дистрофином. По этим причинам каждый человек с МПК должен находиться под наблюдением кардиолога. См. Раздел «Медицинский менеджмент» для получения дополнительной информации о лечении сердечных заболеваний при МПК.
Чтобы просмотреть презентацию кардиолога Элизабет МакНелли о сердце при МПК, см. Видео августа 2012 г. «Сердечные осложнения и лечение при МПК».
Дыхание и кашель
Респираторные мышцы часто остаются сильными при МПК в течение многих лет, но в конечном итоге они могут стать слабее, чем это необходимо для дыхания и кашля (для удаления выделений из дыхательных путей).
Чтобы просмотреть презентацию специалиста по легочной медицине Лизы Вулф в Северо-Западном университете в Чикаго, см. Видео августа 2012 г. «Здоровье легких при нервно-мышечных заболеваниях».
Познание
Врачи считают, что дистрофиновые нарушения в головном мозге могут вызывать когнитивные и поведенческие расстройства и другие психоневрологические расстройства. 6 Умственная отсталость или когнитивные нарушения не являются частыми или тяжелыми у пациентов с диагнозом МПК по сравнению с пациентами с диагнозом МДД. Около 10% пациентов имеют IQ ниже 70. 7,8 Подробнее о том, как справиться с интеллектуальными эффектами, см. Медицинский менеджмент.
Список литературы
- Брэдли, W.Дж., Джонс, М. З., Муссини, Дж. -М, Фосетт, П. Р. У. Мышечная дистрофия по типу Беккера. Мышечный нерв (1978). DOI: 10.1002 / mus.880010204
- Ho, R., Nguyen, M.-L. И Мазер П. Кардиомиопатия при мышечной дистрофии Беккера: Обзор. World J. Cardiol. (2016). DOI: 10.4330 / wjc.v8.i6.356
- Ruiz-Cano, M. J. et al. Успешная трансплантация сердца пациентам с наследственными миопатиями, связанными с терминальной кардиомиопатией. Пересадка.Proc. (2003). DOI: 10.1016 / S0041-1345 (03) 00515-3
- Melacini, P. et al. Поражение миокарда очень часто встречается у пациентов с субклинической мышечной дистрофией Беккера. Тираж (1996). DOI: 10.1161 / 01.CIR.94.12.3168
- Yazawa, M. et al. Семья прогрессирующей мышечной дистрофии Беккера с тяжелой кардиомиопатией. Eur. Neurol. (1987). DOI: 10.1159 / 000116122
- Ricotti, V. et al. Проблемы нервного развития, эмоциональные и поведенческие проблемы при мышечной дистрофии Дюшенна в связи с лежащими в основе мутациями гена дистрофина. Dev. Med. Детский Neurol. (2016). DOI: 10.1111 / dmcn.12922
- Бушби, К. М. и Гарднер-Медвин, Д. Клинические, генетические и дистрофиновые характеристики мышечной дистрофии Беккера — I. Естествознание. J. Neurol. (1993). DOI: 10.1007 / BF00858725
- Daoud, F. et al. Анализ вклада Dp71 в тяжесть умственной отсталости путем сравнения пациентов Дюшенна и Беккера, различающихся по последствиям мутаций на экспрессию Dp71. Гум. Мол. Genet. (2009). DOI: 10.1093 / hmg / ddp320
Симптомы мышечной дистрофии для типов Дюшенна, Беккера и миотонических типов
Дистрофия — это любое состояние, при котором часть тела ослабевает или истощается. При мышечной дистрофии слабость проявляется в мышцах. Унаследованная генетическая ошибка не позволяет организму вырабатывать белок, который помогает наращивать мышцы и сохранять их сильными. Только мужчины могут получить определенные типы мышечной дистрофии из-за того, как это заболевание передается по наследству.
Дети, рожденные с мышечной дистрофией, обычно нормально развиваются в течение первых нескольких лет жизни. Они могут внезапно проявить признаки неуклюжести. К этим признакам относятся:
- проблемы с ходьбой
- трудности с поднятием передней части стопы (так называемое опускание стопы)
- падение
Со временем дети с мышечной дистрофией могут становиться все слабее и слабее, теряя способность сидеть, ходить, и поднимать предметы. Поскольку болезнь также может поражать мышцы сердца и легких, могут возникать одышка и нарушение сердечного ритма.
Существует несколько различных типов мышечной дистрофии. Слабость мышц — отличительный признак каждого типа. Но симптомы могут различаться и проявляться в разном возрасте.
Некоторые мышечные дистрофии легкие. Другие более серьезны и вызывают опасную для жизни мышечную слабость.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — наиболее распространенная и тяжелая форма заболевания. Обычно она начинается в возрасте от 2 до 5 лет.
Симптомы мышечной дистрофии Дюшенна включают:
- Слабость мышц, которая начинается в бедрах, тазе и ногах
- Затруднения при стоянии
- Проблемы с обучением самостоятельно сидеть и ходить
- Неустойчивая походка, ковыляющая
- Ходьба на пальцах ног или подушечках стопы
- Неуклюжесть, частое падение
- Проблемы с подъемом по лестнице
- Затруднения при подъеме из положения лежа или сидя
- Икры больших размеров, иногда болезненные
- Проблемы с дыханием
- Нарушения обучаемости или поведенческие проблемы
- Искривление позвоночника (сколиоз).Это может привести к тому, что одно бедро поднимется выше другого.
- Проблемы с дыханием, которые в конечном итоге могут потребовать использования аппарата ИВЛ.
К 12 годам большинство детей с мышечной дистрофией Дюшенна должны передвигаться в инвалидном кресле. Болезнь также повреждает сердце и мышцы, необходимые для дыхания, что может быть опасно для жизни.
Мышечная дистрофия Беккера
Симптомы мышечной дистрофии Беккера аналогичны симптомам мышечной дистрофии Дюшенна.Но мышечная дистрофия Беккера начинается позже — примерно в подростковом возрасте. Также он развивается намного медленнее.
Продолжение
Первыми признаками мышечной дистрофии Беккера могут быть проблемы с быстрой ходьбой, бегом и подъемом по лестнице. Другие симптомы могут включать:
- Слабость мышц, которая начинается в тазу, плечах, бедрах и бедрах
- Перевязанная походка
- Ходьба на пальцах ног
- Икры больше, чем обычно
- Мышечные судороги при выполнении упражнений
- Проблемы с подъемом предметы выше уровня талии из-за слабости плеч и рук
- Проблемы с сердцем и дыханием (в более зрелом возрасте)
Часто дети с мышечной дистрофией Беккера могут ходить.По мере взросления им может потребоваться трость или инвалидное кресло для передвижения.
Миотоническая дистрофия
Симптомы миотонической дистрофии могут быть очевидны с рождения или развиваться позже — в подростковом или взрослом возрасте.
Как и другие формы мышечной дистрофии, миотоническая дистрофия приводит к мышечной слабости, которая со временем ухудшается, что приводит к неспособности намеренно расслабить мышцы. Обычно сначала поражаются мелкие мышцы, например:
Симптомы миотонической дистрофии могут появиться в любой момент жизни человека.Симптомы включают:
- Слабость мышц лица, рук и шеи
- Жесткость мышц (миотония) — трудности с расслаблением мышц после их напряжения
- Сокращение мышц с течением времени (мышечное истощение)
- Катаракта — помутнение хрусталика глаза
- Дневная сонливость
- Проблемы с обучением и поведением
- Проблемы с сердцем, включая нерегулярное сердцебиение (аритмию)
Тип миотонической дистрофии, которая начинается при рождении, более тяжелый.Другие формы ухудшаются очень медленно, и на прогрессирование может уйти от 50 до 60 лет.
Конечностно-поясная мышечная дистрофия
Эта форма мышечной дистрофии фактически представляет собой группу связанных состояний. Обычно это начинается в детстве или в подростковом возрасте.
Часто мышцы, которые сначала слабеют, оказываются большими мышцами:
Слабость мышц со временем ухудшается очень медленно.
Другие симптомы включают:
- Потеря мышц в пораженных участках
- Боль в спине
- Проблемы с поднятием предметов
- Проблемы с бегом
- Быстрое сердцебиение (учащенное сердцебиение) или нерегулярное сердцебиение
Насколько серьезны последствия, зависит от ребенка .У некоторых детей наблюдается лишь небольшая мышечная слабость. Другие настолько слабы, что им необходимо пользоваться инвалидной коляской.
На более поздних стадиях мышечная дистрофия конечностей и поясов может вызвать серьезные проблемы с сердцем.
Facioscapulohumeral Muscular Dystrophy
Обычно этот тип мышечной дистрофии не проявляется до подросткового возраста или позже. Также очень медленно становится хуже. Некоторые люди могут не осознавать, что у них есть это, пока они не состарятся.
Симптомы включают:
- Слабость мышц лица.Это влияет на способность человека закрывать глаза и поджимать губы (свистеть).
- Слабость мышц в плечах, плечах, верхней части спины и нижних ногах
- Трудности поднимать руки или поднимать предметы из-за мышечной слабости в плечах и спине
Продолжение
Одна сторона тела может быть более серьезной пострадал, чем другой.
Если у вашего ребенка есть симптомы любого типа мышечной дистрофии, обратитесь к педиатру, чтобы узнать, какое дополнительное обследование необходимо.
Мышечная дистрофия — Симптомы и причины
Обзор
Мышечная дистрофия — это группа заболеваний, вызывающих прогрессирующую слабость и потерю мышечной массы. При мышечной дистрофии аномальные гены (мутации) препятствуют выработке белков, необходимых для формирования здоровых мышц.
Есть много видов мышечной дистрофии. Симптомы самого распространенного разнообразия начинаются в детстве, в основном у мальчиков. Другие типы не появляются до зрелого возраста.
Нет лекарства от мышечной дистрофии. Но лекарства и терапия могут помочь справиться с симптомами и замедлить течение болезни.
Продукты и услуги
Показать больше продуктов от Mayo ClinicСимптомы
Основным признаком мышечной дистрофии является прогрессирующая мышечная слабость. Специфические признаки и симптомы начинаются в разном возрасте и в разных группах мышц, в зависимости от типа мышечной дистрофии.
Мышечная дистрофия Дюшенна
Это наиболее распространенная форма. Хотя девочки могут быть переносчиками и иметь легкое поражение, это гораздо чаще встречается у мальчиков.
Признаки и симптомы, которые обычно появляются в раннем детстве, могут включать:
- Частые падения
- Затруднения при вставании из положения лежа или сидя
- Проблемы с бегом и прыжками
- Веревочная походка
- Ходьба на цыпочках
- Большие икроножные мышцы
- Боль и скованность в мышцах
- Нарушения обучаемости
- Отсроченный рост
Мышечная дистрофия Беккера
Признаки и симптомы аналогичны таковым при мышечной дистрофии Дюшенна, но имеют тенденцию быть более легкими и прогрессировать медленнее.Симптомы обычно начинаются в подростковом возрасте, но могут появиться не ранее 20 лет или позже.
Другие типы мышечной дистрофии
Некоторые типы мышечной дистрофии определяются конкретным признаком или местом начала симптомов в организме. Примеры включают:
- Миотонический. Характеризуется неспособностью расслабить мышцы после сокращения. Обычно в первую очередь поражаются мышцы лица и шеи. Люди с этой формой обычно имеют длинные тонкие лица; опущенные веки; и лебединые шеи.
- Facioscapulohumeral (FSHD). Мышечная слабость обычно начинается с лица, бедер и плеч. Лопатки могут выступать, как крылья, когда руки подняты. Начало обычно происходит в подростковом возрасте, но может начаться в детстве или в возрасте 50 лет.
- Врожденный. Этот тип поражает мальчиков и девочек и проявляется при рождении или в возрасте до 2 лет. Некоторые формы прогрессируют медленно и вызывают лишь легкую инвалидность, в то время как другие быстро прогрессируют и вызывают серьезные нарушения.
- Конечностный пояс. Обычно в первую очередь поражаются мышцы бедра и плеча. Люди с этим типом мышечной дистрофии могут испытывать трудности с поднятием передней части стопы и часто спотыкаются. Начало обычно начинается в детстве или подростковом возрасте.
Когда обращаться к врачу
Обратитесь за медицинской помощью, если вы заметили признаки мышечной слабости — например, повышенную неуклюжесть и падение — у вас или вашего ребенка.
Причины
Определенные гены участвуют в производстве белков, защищающих мышечные волокна.Мышечная дистрофия возникает при дефекте одного из этих генов.
Каждая форма мышечной дистрофии вызывается генетической мутацией, характерной для данного типа заболевания. Большинство этих мутаций передаются по наследству.
Факторы риска
Мышечная дистрофия встречается у представителей обоих полов, всех возрастов и рас. Однако наиболее распространенная разновидность, Дюшенн, обычно встречается у мальчиков. Люди с семейным анамнезом мышечной дистрофии подвергаются более высокому риску развития заболевания или передачи его своим детям.
Осложнения
Осложнения прогрессирующей мышечной слабости включают:
- Проблемы при ходьбе. Некоторым людям с мышечной дистрофией со временем необходимо использовать инвалидное кресло.
- Проблемы с использованием оружия. Повседневная деятельность может стать труднее, если затронуты мышцы рук и плеч.
- Укорочение мышц или сухожилий вокруг суставов (контрактуры). Контракты могут еще больше ограничить мобильность.
- Проблемы с дыханием. Прогрессирующая слабость может влиять на мышцы, связанные с дыханием. Людям с мышечной дистрофией, возможно, в конечном итоге потребуется использовать устройство для искусственного дыхания (вентилятор), сначала ночью, но, возможно, также и в течение дня.
- Искривление позвоночника (сколиоз). Ослабленные мышцы могут не удерживать позвоночник прямо.
- Проблемы с сердцем. Мышечная дистрофия может снизить эффективность сердечной мышцы.
- Проблемы с глотанием. При поражении мышц, участвующих в глотании, могут развиться проблемы с питанием и аспирационная пневмония. Питательные трубки могут быть вариантом.
31 января 2020 г.
Симптомы МПК — Новости мышечной дистрофии
Мышечная дистрофия Беккера (МПК) — это наследственное заболевание с истощением мышц, в первую очередь поражающее мальчиков и мужчин.Это вызывает прогрессирующую слабость и истощение скелетных и сердечных мышц.
МПКпохожа на мышечную дистрофию Дюшенна (МДД), но менее распространена и менее выражена. Он ухудшается гораздо медленнее.
Симптомы, которые могут испытывать пациенты с МПК, включают:
Спазмы при физической нагрузке
Начальные симптомы МПК могут включать спазмы и снижение выносливости во время упражнений из-за мышечной слабости в нижних частях тела.
Потеря мышечной массы
Потеря мышечной массы при МПК обычно начинается с бедер, тазовой области, бедер и плеч.Чтобы компенсировать ослабление мышц, пациенты с этим заболеванием могут ходить вразвалкой, ходить на носках или выпирать живот.
Со временем пациенты начинают испытывать трудности при ходьбе, теряют равновесие и координацию, что приводит к частым падениям. У них также могут быть трудности с бегом, прыжками и прыжками, они могут терять мышечную массу и испытывать усталость.
Скорость мышечной дегенерации сильно варьируется от человека к человеку. В то время как некоторым мужчинам требуется инвалидная коляска к 30 годам, другие могут справляться еще много лет с помощью таких вспомогательных средств, как трости.
Сердечные симптомы
BMD может вызвать только легкие проблемы со скелетными мышцами, но серьезные проблемы с сердцем. Слабость сердечной мышцы может привести к одышке, скоплению жидкости в легких и отекам в ступнях и голенях.
Дилатационная кардиомиопатия — сердечная мышца, увеличивающаяся, ослабленная и неспособная эффективно перекачивать кровь — является наиболее частой причиной смерти людей с МПК.
Это делает важным для врачей тщательное наблюдение за пациентами с МПК, чтобы они могли лечить проблемы с сердцем.
Дыхание и кашель
Респираторные мышцы могут оставаться сильными у пациентов с МПК в течение многих лет. Но со временем они могут стать слабее, чем это необходимо для дыхания и кашля, чтобы удалить выделения из дыхательных путей.
Когнитивные проблемы
BMD может вызывать трудности в обучении, которые, по-видимому, возникают в трех основных областях: концентрация внимания, словесное обучение, память и эмоциональное взаимодействие.
Дополнительная информация
Поскольку МПК не влияет напрямую на нервы, осязание и другие чувства остаются в норме, как и контроль пациентов над гладкими или непроизвольными мышцами мочевого пузыря и кишечника и сексуальными функциями.
***
Muscular Dystrophy News — это исключительно новостной и информационный веб-сайт об этом заболевании. Он не предоставляет медицинские консультации, диагностику или лечение. Этот контент не предназначен для замены профессиональных медицинских консультаций, диагностики или лечения. Всегда обращайтесь за советом к своему врачу или другому квалифицированному поставщику медицинских услуг по любым вопросам, которые могут у вас возникнуть относительно состояния здоровья. Никогда не игнорируйте профессиональные медицинские советы и не откладывайте их поиск из-за того, что вы прочитали на этом веб-сайте.
Мышечная дистрофия Дюшенна и Беккера: MedlinePlus Genetics
Мышечные дистрофии — это группа генетических состояний, характеризующихся прогрессирующей мышечной слабостью и истощением (атрофией). Типы мышечной дистрофии Дюшенна и Беккера — это два связанных состояния, которые в первую очередь влияют на скелетные мышцы, которые используются для движения, и сердечную (сердечную) мышцу. Эти формы мышечной дистрофии встречаются почти исключительно у мужчин.
Мышечные дистрофии Дюшенна и Беккера имеют схожие признаки и симптомы и вызываются разными мутациями в одном и том же гене.Эти два состояния различаются по тяжести, возрасту начала и скорости прогрессирования. У мальчиков с мышечной дистрофией Дюшенна мышечная слабость обычно появляется в раннем детстве и быстро ухудшается. У пораженных детей может быть задержка двигательных навыков, таких как сидение, стояние и ходьба. Обычно они становятся инвалидами в подростковом возрасте. Признаки и симптомы мышечной дистрофии Беккера обычно мягче и разнообразнее. В большинстве случаев мышечная слабость проявляется позже в детстве или в подростковом возрасте и ухудшается гораздо медленнее.
Формы мышечной дистрофии Дюшенна и Беккера связаны с сердечным заболеванием, которое называется кардиомиопатией. Эта форма сердечного заболевания ослабляет сердечную мышцу, не позволяя сердцу эффективно перекачивать кровь. При мышечной дистрофии Дюшенна и Беккера кардиомиопатия обычно начинается в подростковом возрасте. Позже сердечная мышца увеличивается, и проблемы с сердцем перерастают в состояние, известное как дилатационная кардиомиопатия. Признаки и симптомы дилатационной кардиомиопатии могут включать нерегулярное сердцебиение (аритмию), одышку, сильную усталость (утомляемость) и отек ног и ступней.Эти проблемы с сердцем быстро ухудшаются и в большинстве случаев становятся опасными для жизни. Мужчины с мышечной дистрофией Дюшенна обычно доживают до двадцати лет, в то время как мужчины с мышечной дистрофией Беккера могут дожить до сорока лет и старше.
Родственное состояние, называемое Х-сцепленной дилатационной кардиомиопатией, представляет собой форму сердечного заболевания, вызванную мутациями в том же гене, что и мышечная дистрофия Дюшенна и Беккера, и иногда его классифицируют как субклиническую мышечную дистрофию Беккера. Люди с Х-сцепленной дилатационной кардиомиопатией обычно не имеют слабости или истощения скелетных мышц, хотя у них могут быть незначительные изменения в клетках скелетных мышц, которые можно обнаружить с помощью лабораторных исследований.
Мышечная дистрофия Беккера: основы практики, патофизиология, эпидемиология
Беккер П.Е., Кинер Ф. [Новая х-хромосомная мышечная дистрофия.]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr . 1955. 193 (4): 427-48. [Медлайн].
Becker PE. Два семейства доброкачественной рецессивной мышечной дистрофии, связанной с полом. Рев. Банка Биол . 1962 сентябрь-декабрь. 21: 551-66. [Медлайн].
Andrews JG, Wahl RA.Мышечная дистрофия Дюшенна и Беккера у подростков: современные перспективы. Мед. Здоровья подростков Ther . 2018. 9: 53-63. [Медлайн]. [Полный текст].
Thada PK, Bhandari J, Umapathi KK. Мышечная дистрофия Беккера. StatPearls . 2021 Январь [Medline]. [Полный текст].
Angelini C, Fanin M, Pegoraro E, et al. Клинико-молекулярная корреляция у 104 пациентов с легкой Х-сцепленной мышечной дистрофией: характеристика субклинических фенотипов. Нервно-мышечное расстройство . 1994 июл.4 (4): 349-58. [Медлайн].
Гурвич О.Л., Туохи Т.М., Ховард М.Т. и др. Мутации псевдоэксона МДД: эффективность сплайсинга, фенотип и потенциальная терапия. Энн Нейрол . 2008, январь, 63 (1): 81-9. [Медлайн].
Эштон Э.Дж., Яу С.К., Динз З.С. и др. Одновременное сканирование мутаций на предмет грубых делеций, дупликаций и точечных мутаций в гене DMD. евро J Hum Genet . 2008 16 января (1): 53-61.[Медлайн].
Arahata K, Beggs AH, Honda H и др. Сохранение С-конца молекулы дистрофина в скелетных мышцах от мышечной дистрофии Беккера. J Neurol Sci . 1991 Февраль 101 (2): 148-56. [Медлайн].
Кениг М., Беггс А.Х., Мойер М. и др. Молекулярная основа мышечной дистрофии Дюшенна и Беккера: корреляция степени тяжести с типом делеции. Ам Дж. Хам Генет . 1989 Октябрь 45 (4): 498-506. [Медлайн].[Полный текст].
Schwartz M, Hertz JM, Sveen ML, et al. LGMD2I с характерным фенотипом мышечной дистрофии Дюшенна или Беккера. Неврология . 2005 10 мая. 64 (9): 1635-7. [Медлайн].
Cirak S, Feng L, Anthony K, Arechavala-Gomeza V, Torelli S, Sewry C и др. Восстановление дистрофин-ассоциированного гликопротеинового комплекса после терапии пропуска экзонов при мышечной дистрофии Дюшенна. Мол тер . 2011 15 ноя.[Медлайн].
Mavrogeni S, Markousis-Mavrogenis G, Papavasiliou A, Kolovou G. Поражение сердца при мышечной дистрофии Дюшенна и Беккера. Мир Дж. Кардиол . 2015 26 июля. 7 (7): 410-4. [Медлайн].
Николас А., Рагуэн-Николь С., Бен Яу Р. и др. Выраженность мышечной дистрофии Беккера связана со структурой дистрофина. Хум Мол Генет . 2015 г., 1. 24 (5): 1267-79. [Медлайн].
Наблюдение за здоровьем сердечно-сосудистой системы для лиц, страдающих мышечной дистрофией Дюшенна или Беккера. Педиатрия . 2005 декабрь 116 (6): 1569-73. [Медлайн]. [Полный текст].
Эмери А.Е., Скиннер Р. Клинические исследования доброкачественной (тип Беккера) Х-сцепленной мышечной дистрофии. Клин Генет . 1976 Октябрь 10 (4): 189-201. [Медлайн].
Holloway SM, Wilcox DE, Wilcox A, et al. Ожидаемая продолжительность жизни и смерть от кардиомиопатии среди носителей мышечной дистрофии Дюшенна и Беккера в Шотландии. Сердце . 11 октября 2007 г. [Medline].
Young HK, Barton BA, Waisbren S, et al. Когнитивный и психологический профиль мужчин с мышечной дистрофией Беккера. J Детский нейрол . 2007 г. 3 декабря [Medline].
De Wel B, Willaert S, Nadaj-Pakleza A, et al. Снижение дыхания у взрослых пациентов с мышечной дистрофией Беккера: продольное исследование. Нервно-мышечное расстройство . 2020 26 декабря. [Medline].
Zhang H, Zhu Y, Sun Y, et al.Уровень креатинина в сыворотке: дополнительный показатель, позволяющий отличить мышечную дистрофию Дюшенна от мышечной дистрофии Беккера. Маркер Дис . 2015. 2015: 141856. [Медлайн].
Menezes MP, North KN. Унаследованные нервно-мышечные расстройства: путь к диагностике. J Детский педиатр . 2011 г. 3 ноября [Medline].
Лим BC, Ли С., Шин Дж.Й., Ким Джи, Хван Х, Ким К.Дж. и др. Генетическая диагностика мышечной дистрофии Дюшенна и Беккера с использованием технологии секвенирования нового поколения: комплексный поиск мутаций на единой платформе. Дж Мед Генет . 2011 ноябрь 48 (11): 731-6. [Медлайн].
Марти Б., Жиль Р., Туссент М. и др. Комплексная оценка структурных и функциональных нарушений миокарда при мышечной дистрофии Беккера с использованием количественной магнитно-резонансной томографии сердца. Eur Heart J Cardiovasc Imaging . 24 декабря 2018 г. [Medline].
Руткове С.Б., Даррас БТ. Электроимпедансная миография для оценки детей с мышечной дистрофией: предварительное исследование. Журнал Физической конференции, сер. . 2013. 434 (1): [Medline]. [Полный текст].
Grootenhuis MA, de Boone J, van der Kooi AJ. Жизнь с мышечной дистрофией: последствия для качества жизни, связанные со здоровьем, для детей и взрослых. Результаты здорового образа жизни . 2007. 5:31. [Медлайн]. [Полный текст].
Ямада Ю., Каваками М., Вада А., Оцука Т., Мураока К., Лю М. Сравнение дисфункции глотания при мышечной дистрофии Беккера и мышечной дистрофии Дюшенна. Дезабил Рехабил . 2017 13 марта. 1-5. [Медлайн].
Hayes J, Veyckemans F, Bissonnette B. Мышечная дистрофия Дюшенна: возвращение к старой проблеме анестезии. Педиатр Анаэст . 2008 18 февраля (2): 100-6. [Медлайн].
Segura LG, Lorenz JD, Weingarten TN, Scavonetto F, Bojanic K, Selcen D, et al. Анестезия и мышечная дистрофия Дюшенна или Беккера: обзор 117 воздействий анестетика. Педиатр Анаэст .2013 Сентябрь 23 (9): 855-64. [Медлайн].
Дженкинс Х.М., Стоки А., Криеллаарс Д., Пастеркамп Х. Накладывание дыхания у детей с нервно-мышечными расстройствами. Пульмонол Педиатр . 2013 16 августа [Medline].
Pouwels S, de Boer A, Leufkens HG, Weber WE, Cooper C, van Onzenoort HA, et al. Риск перелома у пациентов с мышечными дистрофиями. Остеопорос Инт . 2013 15 августа [Medline].
Дуан Д.Myodys, полноразмерный плазмидный вектор дистрофина для генной терапии мышечной дистрофии Дюшенна и Беккера.